Functional and Biochemical Consequences of Disease Variants in Neurotransmitter Transporters: a Special Emphasis on Folding and Trafficking Deficits
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table S1 the Four Gene Sets Derived from Gene Expression Profiles of Escs and Differentiated Cells
Table S1 The four gene sets derived from gene expression profiles of ESCs and differentiated cells Uniform High Uniform Low ES Up ES Down EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol 269261 Rpl12 11354 Abpa 68239 Krt42 15132 Hbb-bh1 67891 Rpl4 11537 Cfd 26380 Esrrb 15126 Hba-x 55949 Eef1b2 11698 Ambn 73703 Dppa2 15111 Hand2 18148 Npm1 11730 Ang3 67374 Jam2 65255 Asb4 67427 Rps20 11731 Ang2 22702 Zfp42 17292 Mesp1 15481 Hspa8 11807 Apoa2 58865 Tdh 19737 Rgs5 100041686 LOC100041686 11814 Apoc3 26388 Ifi202b 225518 Prdm6 11983 Atpif1 11945 Atp4b 11614 Nr0b1 20378 Frzb 19241 Tmsb4x 12007 Azgp1 76815 Calcoco2 12767 Cxcr4 20116 Rps8 12044 Bcl2a1a 219132 D14Ertd668e 103889 Hoxb2 20103 Rps5 12047 Bcl2a1d 381411 Gm1967 17701 Msx1 14694 Gnb2l1 12049 Bcl2l10 20899 Stra8 23796 Aplnr 19941 Rpl26 12096 Bglap1 78625 1700061G19Rik 12627 Cfc1 12070 Ngfrap1 12097 Bglap2 21816 Tgm1 12622 Cer1 19989 Rpl7 12267 C3ar1 67405 Nts 21385 Tbx2 19896 Rpl10a 12279 C9 435337 EG435337 56720 Tdo2 20044 Rps14 12391 Cav3 545913 Zscan4d 16869 Lhx1 19175 Psmb6 12409 Cbr2 244448 Triml1 22253 Unc5c 22627 Ywhae 12477 Ctla4 69134 2200001I15Rik 14174 Fgf3 19951 Rpl32 12523 Cd84 66065 Hsd17b14 16542 Kdr 66152 1110020P15Rik 12524 Cd86 81879 Tcfcp2l1 15122 Hba-a1 66489 Rpl35 12640 Cga 17907 Mylpf 15414 Hoxb6 15519 Hsp90aa1 12642 Ch25h 26424 Nr5a2 210530 Leprel1 66483 Rpl36al 12655 Chi3l3 83560 Tex14 12338 Capn6 27370 Rps26 12796 Camp 17450 Morc1 20671 Sox17 66576 Uqcrh 12869 Cox8b 79455 Pdcl2 20613 Snai1 22154 Tubb5 12959 Cryba4 231821 Centa1 17897 -

Identification of a Novel Nucleobase-Ascorbate Transporter
bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Identification of a novel Nucleobase-Ascorbate 2 Transporter family member in fish and amphibians 3 Diogo Oliveira*,1, André M. Machado*,1, Tiago Cardoso1, Mónica Lopes-Marques1, L. Filipe 4 C. Castro1,2● and Raquel Ruivo1● 5 1CIIMAR – Interdisciplinary Centre of Marine and Environmental Research, U. Porto – University 6 of Porto, Porto, Portugal 7 2Department of Biology, Faculty of Sciences, U. Porto - University of Porto, Portugal 8 *Equal contribution 9 ●Corresponding authors at: CIIMAR, Terminal de Cruzeiros do Porto de Leixões, Av. General 10 Norton de Matos s/n, 4450-208 Matosinhos, Portugal. Tel.: +351 223 401 831 11 12 Running title: Novel uric acid transporter in fish and amphibians 13 14 15 bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 16 Abstract:Nucleobase-Ascorbate Transporter (NAT) family includes ascorbic acid, nucleobases 17 and uric acid transporters: with a broad evolutionary distribution. In vertebrates, four members 18 have been previously recognized, the ascorbate transporters Slc23a1 and Slc3a2, the nucleobase 19 transporter Slc23a4 and an orphan transporter Slc23a3. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
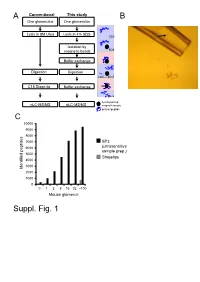
Suppl. Fig. 1 A
A Conventional This study B One glomerulus One glomerulus Lysis in 8M Urea Lysis in 4% SDS SDS Isolation by magnetic beads SDS Buffer exchange Digestion Digestion Try LysC pH>7 C18 Stage tip Buffer exchange pH<4 functionalized nLC-MS/MS nLC-MS/MS magnetic beads protein/peptide C 10000 9000 8000 7000 SP3 6000 (ultrasensitive sample prep.) 5000 Stagetips 4000 3000 Identified peptides 2000 1000 0 0 1 2 8 16 32 ~100 Mouse glomeruli Suppl. Fig. 1 S1. Sample preparation methods. A. Scheme of conventional (C18 Stage Tip) and ultrasensitive proteomic sample preparation methods. B. Manual microdissection and isolation of single glomeruli. The arrow indicates a single isolated glomerulus subjected to proteome analysis. The tip capacity is 10µl. C. Stagetips, a conventional proteomic sample preparation method, is compared with ultrasensitive (SP3) proteome analysis. The indicated amounts of mouse glomeruli were prepared by the respective sample preparation protocols, and peptide numbers after nLC-MS/MS are plotted (all FDR<0.01). A 100 1 Tubule B 80 60 40 Intensity CCD 20 40 0 100 20 S1 TAL 80 0 Tubule proximal tubule Loading... 60 0 Intensity 40 S2 20 -20 control Component 2 (22.5%) S3 0 0 10 20 30 40 50 60 -40 -20 0 20 40 Time (min) Component 2 (47.1%) C D 6000 Mouse proximal tubule 5000 8 4000 3000 7 (iBAQ) Loading... 10 2000 log 6 Number of peptides 1000 0 5 Control mouse mouse mouse human proximal mTAL CCD proximal 0 1500 tubule tubule Rank protein (S1) (S1) R=0.7 E F 8 ACTB Human proximal tubule 8 7 SLC9A3R1 ATP1A1 SLC3A2 SLC25A5 ATP1B1 7 SLC5A2 SLC25A3 CUBN CLTC SLC27A2 SLC5A12 LRP2 6 SLC25A13 SLC25A10 SLC5A1 SLC22A6 6 SLC25A4 Loading.. -

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -

Genes in Eyecare Geneseyedoc 3 W.M
Genes in Eyecare geneseyedoc 3 W.M. Lyle and T.D. Williams 15 Mar 04 This information has been gathered from several sources; however, the principal source is V. A. McKusick’s Mendelian Inheritance in Man on CD-ROM. Baltimore, Johns Hopkins University Press, 1998. Other sources include McKusick’s, Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore. Johns Hopkins University Press 1998 (12th edition). http://www.ncbi.nlm.nih.gov/Omim See also S.P.Daiger, L.S. Sullivan, and B.J.F. Rossiter Ret Net http://www.sph.uth.tmc.edu/Retnet disease.htm/. Also E.I. Traboulsi’s, Genetic Diseases of the Eye, New York, Oxford University Press, 1998. And Genetics in Primary Eyecare and Clinical Medicine by M.R. Seashore and R.S.Wappner, Appleton and Lange 1996. M. Ridley’s book Genome published in 2000 by Perennial provides additional information. Ridley estimates that we have 60,000 to 80,000 genes. See also R.M. Henig’s book The Monk in the Garden: The Lost and Found Genius of Gregor Mendel, published by Houghton Mifflin in 2001 which tells about the Father of Genetics. The 3rd edition of F. H. Roy’s book Ocular Syndromes and Systemic Diseases published by Lippincott Williams & Wilkins in 2002 facilitates differential diagnosis. Additional information is provided in D. Pavan-Langston’s Manual of Ocular Diagnosis and Therapy (5th edition) published by Lippincott Williams & Wilkins in 2002. M.A. Foote wrote Basic Human Genetics for Medical Writers in the AMWA Journal 2002;17:7-17. A compilation such as this might suggest that one gene = one disease. -

Towards the Elucidation of Orphan Lysosomal Transporters Quentin Verdon
Towards the Elucidation of Orphan Lysosomal Transporters Quentin Verdon To cite this version: Quentin Verdon. Towards the Elucidation of Orphan Lysosomal Transporters. Cancer. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS144. tel-01827233 HAL Id: tel-01827233 https://tel.archives-ouvertes.fr/tel-01827233 Submitted on 2 Jul 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS144 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY PREPAREE A L’UNIVERSITE PARIS-SUD ECOLE DOCTORALE N°568 BIOSIGNE | Signalisations et réseaux intégratifs en biologie Spécialité de doctorat : aspects moléculaires et cellulaires de la biologie Par Mr Quentin Verdon Towards the elucidation of orphan lysosomal transporters: several shots on target and one goal Thèse présentée et soutenue à Paris le 29/06/2016 » : Composition du Jury : Mr Le Maire Marc Professeur, Université Paris-Sud Président Mr Birman Serge Directeur de recherche, CNRS Rapporteur Mr Murray James Assistant professor, Trinity college Dublin Rapporteur Mr Goud Bruno Directeur de recherche, CNRS Examinateur Mr Gasnier Bruno Directeur de recherche, CNRS Directeur de thèse Mme Sagné Corinne Chargée de recherche, INSERM Co-directeur de thèse Table of contents Remerciements (acknowledgements) 6 Abbreviations 7 Abstracts 10 Introduction 12 1 Physiology of lysosomes 12 1.1 Discovery and generalities 12 1.2 Degradative function 13 1.3. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Biomarker Status of the Human Equilibrative Nucleoside
cancers Article “Open Sesame?”: Biomarker Status of the Human Equilibrative Nucleoside Transporter-1 and Molecular Mechanisms Influencing its Expression and Activity in the Uptake and Cytotoxicity of Gemcitabine in Pancreatic Cancer 1,2, 1, 1,3 1 Ornella Randazzo y, Filippo Papini y, Giulia Mantini , Alessandro Gregori , Barbara Parrino 2, Daniel S. K. Liu 4 , Stella Cascioferro 2 , Daniela Carbone 2 , 1,5 4,6, 1,7, Godefridus J. Peters , Adam E. Frampton * , Ingrid Garajova y and Elisa Giovannetti 1,3,* 1 Department of Medical Oncology, Cancer Center Amsterdam, Amsterdam UMC, VU University Medical Center (VUmc), 1081 HV Amsterdam, The Netherlands; [email protected] (O.R.); [email protected] (F.P.); [email protected] (G.M.); [email protected] (A.G.); [email protected] (G.J.P.); [email protected] (I.G.) 2 Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche (STEBICEF), Università degli Studi di Palermo, 90123 Palermo, Italy; [email protected] (B.P.); [email protected] (S.C.); [email protected] (D.C.) 3 Cancer Pharmacology Lab, AIRC Start Up Unit, Fondazione Pisana per la Scienza, 56017 Pisa, Italy 4 Division of Cancer, Department of Surgery & Cancer, Imperial College, Hammersmith Hospital campus, London W12 0NN, UK;; [email protected] 5 Department of Biochemistry, Medical University of Gdansk, 80-210 Gdansk, Poland 6 Faculty of Health and Medical Sciences, The Leggett Building, University of Surrey, Guildford GU2 7XH, UK 7 Medical Oncology Unit, University Hospital of Parma, Via Gramsci 14, 43126 Parma, Italy * Correspondence: [email protected] (A.E.F.); [email protected] (E.G.); Tel.: +31-003-120-444-2633 (E.G.) These authors contributed equally to this paper. -

Toxicological Profile for Nitrate and Nitrite
NITRATE AND NITRITE 29 3. HEALTH EFFECTS 3.1 INTRODUCTION The primary purpose of this chapter is to provide public health officials, physicians, toxicologists, and other interested individuals and groups with an overall perspective on the toxicology of nitrate and nitrite. It contains descriptions and evaluations of toxicological studies and epidemiological investigations and provides conclusions, where possible, on the relevance of toxicity and toxicokinetic data to public health. A glossary and list of acronyms, abbreviations, and symbols can be found at the end of this profile. 3.2 DISCUSSION OF HEALTH EFFECTS BY ROUTE OF EXPOSURE To help public health professionals and others address the needs of persons living or working near hazardous waste sites, the information in this section is organized first by route of exposure (inhalation, oral, and dermal) and then by health effect (e.g., death, systemic, immunological, neurological, reproductive, developmental, and carcinogenic effects). These data are discussed in terms of three exposure periods: acute (14 days or less), intermediate (15–364 days), and chronic (365 days or more). Levels of significant exposure for each route and duration are presented in tables and illustrated in figures. The points in the figures showing no-observed-adverse-effect levels (NOAELs) or lowest- observed-adverse-effect levels (LOAELs) reflect the actual doses (levels of exposure) used in the studies. LOAELs have been classified into "less serious" or "serious" effects. "Serious" effects are those that evoke failure in a biological system and can lead to morbidity or mortality (e.g., acute respiratory distress or death). "Less serious" effects are those that are not expected to cause significant dysfunction or death, or those whose significance to the organism is not entirely clear. -

Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb As an Allosteric Modulator of the Human Dopamine Transporter
biomedicines Article Functional Characterization of the Dopaminergic Psychostimulant Sydnocarb as an Allosteric Modulator of the Human Dopamine Transporter Shaili Aggarwal 1, Mary Hongying Cheng 2 , Joseph M. Salvino 3 , Ivet Bahar 2 and Ole Valente Mortensen 1,* 1 Department of Pharmacology and Physiology, Drexel University College of Medicine, Philadelphia, PA 19102, USA; [email protected] 2 Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15260, USA; [email protected] (M.H.C.); [email protected] (I.B.) 3 The Wistar Institute, Philadelphia, PA 19104, USA; [email protected] * Correspondence: [email protected] Abstract: The dopamine transporter (DAT) serves a critical role in controlling dopamine (DA)- mediated neurotransmission by regulating the clearance of DA from the synapse and extrasynaptic regions and thereby modulating DA action at postsynaptic DA receptors. Major drugs of abuse such as amphetamine and cocaine interact with DATs to alter their actions resulting in an enhancement in extracellular DA concentrations. We previously identified a novel allosteric site in the DAT and the related human serotonin transporter that lies outside the central orthosteric substrate- and cocaine-binding pocket. Here, we demonstrate that the dopaminergic psychostimulant sydnocarb is a ligand of this novel allosteric site. We identified the molecular determinants of the interaction between sydnocarb and DAT at the allosteric site using molecular dynamics simulations. Biochemical- Citation: Aggarwal, S.; Cheng, M.H.; Salvino, J.M.; Bahar, I.; Mortensen, substituted cysteine scanning accessibility experiments have supported the computational predictions O.V. Functional Characterization of by demonstrating the occurrence of specific interactions between sydnocarb and amino acids within the Dopaminergic Psychostimulant the allosteric site. -

Compositions and Methods for Selective Delivery of Oligonucleotide Molecules to Specific Neuron Types
(19) TZZ ¥Z_T (11) EP 2 380 595 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.10.2011 Bulletin 2011/43 A61K 47/48 (2006.01) C12N 15/11 (2006.01) A61P 25/00 (2006.01) A61K 49/00 (2006.01) (2006.01) (21) Application number: 10382087.4 A61K 51/00 (22) Date of filing: 19.04.2010 (84) Designated Contracting States: • Alvarado Urbina, Gabriel AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Nepean Ontario K2G 4Z1 (CA) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • Bortolozzi Biassoni, Analia Alejandra PT RO SE SI SK SM TR E-08036, Barcelona (ES) Designated Extension States: • Artigas Perez, Francesc AL BA ME RS E-08036, Barcelona (ES) • Vila Bover, Miquel (71) Applicant: Nlife Therapeutics S.L. 15006 La Coruna (ES) E-08035, Barcelona (ES) (72) Inventors: (74) Representative: ABG Patentes, S.L. • Montefeltro, Andrés Pablo Avenida de Burgos 16D E-08014, Barcelon (ES) Edificio Euromor 28036 Madrid (ES) (54) Compositions and methods for selective delivery of oligonucleotide molecules to specific neuron types (57) The invention provides a conjugate comprising nucleuc acid toi cell of interests and thus, for the treat- (i) a nucleic acid which is complementary to a target nu- ment of diseases which require a down-regulation of the cleic acid sequence and which expression prevents or protein encoded by the target nucleic acid as well as for reduces expression of the target nucleic acid and (ii) a the delivery of contrast agents to the cells for diagnostic selectivity agent which is capable of binding with high purposes.