BRAF Controls the Effects of Metformin on Neuroblast Cell Divisions in C. Elegans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
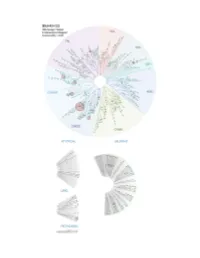
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Protein Kinase C As a Paradigm Alexandra C
Biochem. J. (2003) 370, 361–371 (Printed in Great Britain) 361 REVIEW ARTICLE Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm Alexandra C. NEWTON1 Department of Pharmacology, University of California at San Diego, La Jolla, CA 92093-0640, U.S.A. Phosphorylation plays a central role in regulating the activation down-regulation of PKC as a paradigm for how these sites and signalling lifetime of protein kinases A, B (also known as control the function of the ABC kinases. Akt) and C. These kinases share three conserved phosphorylation motifs: the activation loop segment, the turn motif and the Key words: phosphoinositide 3-kinase, phosphoinositide-depen- hydrophobic motif. This review focuses on how phosphorylation dent kinase-1 (PDK-1), phosphorylation motif, protein kinase A at each of these sites regulates the maturation, signalling and (PKA), protein kinase B (PKB)\Akt, protein kinase C (PKC). INTRODUCTION by phosphorylation as a paradigm for control of ABC kinase function by phosphorylation. Almost half a century ago Krebs, Graves and Fischer reported b that the first discovered protein kinase, phosphorylase kinase, ARCHITECTURE OF ABC KINASES was, itself, activated by phosphorylation [1]. Two decades later, Fischer and colleagues showed that the kinase that phosphory- Protein kinases A, B\Akt and C have in common a conserved lates phosphorylase kinase, later named protein kinase A (PKA), kinase core whose function is regulated allosterically by a was also phosphorylated [2]. Reversible control by phosphory- corresponding regulatory moiety (Figure 1). In the case of PKA, lation\dephosphorylation is now firmly established as a fun- the regulatory moiety is a separate polypeptide, whereas the damental mechanism for regulating the function of most of the regulatory determinants are on the same polypeptide as the 500 or so members of the mammalian protein kinase superfamily. -

And Glucocorticoid-Inducible Kinase (Sgk1)" (2007)
Dartmouth College Dartmouth Digital Commons Open Dartmouth: Published works by Dartmouth faculty Faculty Work 2-14-2007 Regulation of Human Cystic Fibrosis Transmembrane Conductance Regulator (Cftr) by Serum- and Glucocorticoid- Inducible Kinase (Sgk1) J. Denry Sato Mount Desert Island Biological Laboratory, Salisbury Cove M. Christine Chapline Dartmouth College Renee Thibodeau Dartmouth College Raymond A. Frizzell University of Pittsburgh Bruce A. Stanton Dartmouth College Follow this and additional works at: https://digitalcommons.dartmouth.edu/facoa Part of the Medicine and Health Sciences Commons Dartmouth Digital Commons Citation Sato, J. Denry; Chapline, M. Christine; Thibodeau, Renee; Frizzell, Raymond A.; and Stanton, Bruce A., "Regulation of Human Cystic Fibrosis Transmembrane Conductance Regulator (Cftr) by Serum- and Glucocorticoid-Inducible Kinase (Sgk1)" (2007). Open Dartmouth: Published works by Dartmouth faculty. 3156. https://digitalcommons.dartmouth.edu/facoa/3156 This Article is brought to you for free and open access by the Faculty Work at Dartmouth Digital Commons. It has been accepted for inclusion in Open Dartmouth: Published works by Dartmouth faculty by an authorized administrator of Dartmouth Digital Commons. For more information, please contact [email protected]. Original Paper Cellular Physiology Cell Physiol Biochem 2007;20:91-98 Accepted: February 14, 2007 and Biochemistry Regulation of Human Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) by Serum- and Glucocorticoid-Inducible Kinase (SGK1) J. Denry Sato1, M. Christine Chapline1,2, Renee Thibodeau1,2, Raymond A. Frizzell1,3 and Bruce A. Stanton1,2 1Mount Desert Island Biological Laboratory, Salisbury Cove, 2Dept. of Physiology, Dartmouth Medical School, Hanover, 3Dept. of Cell Biology and Physiology, University of Pittsburgh Medical School, Pittsburgh Key Words in stimulating CFTR Cl currents. -

Mtor Signaling in Metabolism and Cancer
cells Editorial mTOR Signaling in Metabolism and Cancer Shile Huang 1,2 1 Department of Biochemistry and Molecular Biology, Louisiana State University Health Sciences Center, 1501 Kings Highway, Shreveport, LA 71130-3932, USA; [email protected]; Tel.: +1-318-675-7759 2 Feist-Weiller Cancer Center, Louisiana State University Health Sciences Center, 1501 Kings Highway, Shreveport, LA 71130-3932, USA Received: 10 October 2020; Accepted: 13 October 2020; Published: 13 October 2020 Abstract: The mechanistic/mammalian target of rapamycin (mTOR), a serine/threonine kinase, is a central regulator for human physiological activity. Deregulated mTOR signaling is implicated in a variety of disorders, such as cancer, obesity, diabetes, and neurodegenerative diseases. The papers published in this special issue summarize the current understanding of the mTOR pathway and its role in the regulation of tissue regeneration, regulatory T cell differentiation and function, and different types of cancer including hematologic malignancies, skin, prostate, breast, and head and neck cancer. The findings highlight that targeting the mTOR pathway is a promising strategy to fight against certain human diseases. Keywords: mTOR; PI3K; Akt; tissue regeneration; regulatory T cells; tumor; photodynamic therapy The mechanistic/mammalian target of rapamycin (mTOR), a serine/threonine kinase, integrates environmental cues such as hormones, growth factors, nutrients, oxygen, and energy, regulating cell growth, proliferation, survival, motility and differentiation as well as metabolism (reviewed in [1,2]). Evidence has demonstrated that deregulated mTOR signaling is implicated in a variety of disorders, such as cancer, obesity, diabetes, and neurodegenerative diseases (reviewed in [1,2]). Current knowledge indicates that mTOR functions at least as two distinct complexes (mTORC1 and mTORC2) in mammalian cells. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

Modulation of TORC2 Signaling by a Conserved Lkb1 Signaling Axis in Budding Yeast
| INVESTIGATION Modulation of TORC2 Signaling by a Conserved Lkb1 Signaling Axis in Budding Yeast Maria Alcaide-Gavilán,*,1 Rafael Lucena,*,1,2 Katherine A. Schubert,* Karen L. Artiles,*,3 Jessica Zapata,*,†,4 and Douglas R. Kellogg*,2 *Department of Molecular, Cell, and Developmental Biology, University of California, Santa Cruz, California 95064 and †NantOmics, Santa Cruz, CA 95064 ORCID IDs: 0000-0001-9102-4857 (M.A.-G.); 0000-0003-2050-0611 (R.L.); 0000-0003-2676-8838 (K.A.S.); 0000-0002-1674-064X (K.L.A.); 0000-0001-9567-3539 (J.Z.); 0000-0002-5050-2194 (D.R.K.) ABSTRACT Nutrient availability, growth rate, and cell size are closely linked. For example, in budding yeast, the rate of cell growth is proportional to nutrient availability, cell size is proportional to growth rate, and growth rate is proportional to cell size. Thus, cells grow slowly in poor nutrients and are nearly half the size of cells growing in rich nutrients. Moreover, large cells grow faster than small cells. A signaling network that surrounds TOR kinase complex 2 (TORC2) plays an important role in enforcing these proportional relationships. Cells that lack components of the TORC2 network fail to modulate their growth rate or size in response to changes in nutrient availability. Here, we show that budding yeast homologs of the Lkb1 tumor suppressor kinase are required for normal modulation of TORC2 signaling in response to changes in carbon source. Lkb1 kinases activate Snf1/AMPK to initiate transcription of genes required for utilization of poor carbon sources. However, Lkb1 influences TORC2 signaling via a novel pathway that is independent of Snf1/AMPK. -

Balancing Mtor Signaling and Autophagy in the Treatment of Parkinson's Disease
Review Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease Zhou Zhu 1 , Chuanbin Yang 1, Ashok Iyaswamy 1, Senthilkumar Krishnamoorthi 1, Sravan Gopalkrishnashetty Sreenivasmurthy 1, Jia Liu 1, Ziying Wang 1, Benjamin Chun-Kit Tong 1 , Juxian Song 2, Jiahong Lu 3, King-Ho Cheung 1 and Min Li 1,* 1 Mr. and Mrs. Ko Chi Ming Centre for Parkinson’s Disease Research, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong SAR 999077, China; [email protected] (Z.Z.); [email protected] (C.Y.); [email protected] (A.I.); [email protected] (S.K.); [email protected] (S.G.S.); [email protected] (J.L.); [email protected] (Z.W.); [email protected] (B.C.-K.T.); [email protected] (K.-H.C.) 2 Medical College of Acupuncture-Moxibustion and Rehabilitation, Guangzhou University of Chinese Medicine, Guangzhou 510006, China; [email protected] 3 State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Taipa, Macau SAR 999078, China; [email protected] * Correspondence: [email protected]; Tel: +852-3411-2919 Received: 15 January 2019; Accepted: 1 February 2019; Published: 8 February 2019 Abstract: The mammalian target of rapamycin (mTOR) signaling pathway plays a critical role in regulating cell growth, proliferation, and life span. mTOR signaling is a central regulator of autophagy by modulating multiple aspects of the autophagy process, such as initiation, process, and termination through controlling the activity of the unc51-like kinase 1 (ULK1) complex and vacuolar protein sorting 34 (VPS34) complex, and the intracellular distribution of TFEB/TFE3 and proto-lysosome tubule reformation. -

SGK Antibody A
C 0 2 - t SGK Antibody a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 2 7 Web: [email protected] 2 www.cellsignal.com 3 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source: UniProt ID: Entrez-Gene Id: WB H Transfected Only 54 (Transfected Rabbit O00141 6446 only) Product Usage Information Application Dilution Western Blotting 1:1000 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA and 50% glycerol. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity SGK Antibody detects transfected levels of SGK1, SGK2 and SGK3. Species Reactivity: Human Species predicted to react based on 100% sequence homology: Mouse, Rat Source / Purification Polyclonal antibodies are produced by immunizing animals with a synthetic peptide corresponding to residues surrounding the carboxy-terminal domain of human SGK. Antibodies are purified by protein A and peptide affinity chromatography. Background Serum and glucocorticoid-inducible kinase (SGK) is a serine/threonine kinase closely related to Akt (1). SGK is rapidly induced in response to a variety of stimuli, including serum, glucocorticoid, follicle stimulating hormone, osmotic shock, and mineralocorticoids. SGK activation can be accomplished via HGF PI3K-dependent pathways and by integrin- mediated PI3K-independent pathways (2,3). Induction and activation of SGK has been implicated in activating the modulation of anti-apoptotic and cell cycle regulation (4-6). SGK also plays an important role in activating certain potassium, sodium, and chloride channels, suggesting its involvement in the regulation of processes such as cell survival, neuronal excitability, and renal sodium excretion (2). -

SGK2 Active Human (S7570)
SGK2, Active Human, recombinant, expressed in E. coli Product Number S 7570 Storage Temperature: -70 °C Synonym: Serum/Glucocorticoid-Regulated Kinase 2 Purity: ³ 75% (SDS-PAGE) Product Description Molecular weight: ~66 kDa SGK2 is a member of the serum- and glucocorticoid- induced kinases (SGK) which are serine-threonine Specific Activity: ³ 100 units/mg protein (Bradford). kinases and belong to the "AGC" kinase subfamily, Please refer to the Certificate of Analysis for the which includes protein kinases A, G, and C. The SGK2 lot-specific activity. catalytic domain is most similar to protein kinase B 1 (PKB). SGK1 was originally identified as a Unit Definition: One unit will incorporate one nanomole glucocorticoid-sensitive gene and, subsequently, the of phosphate into the Akt/SGK substrate peptide homologous kinases SGK2 and SGK3 have been (RPRAATF) per minute at 30 °C at pH 7.2 using a cloned. They are products of distinct genes, which are final concentration of 50 µM [32P] ATP. differentially expressed and share 80% identity in their 2 catalytic domains. SGK2, like SGK1 and SGK3, is Precautions and Disclaimer stimulated by insulin and insulin-like growth factor-1 This product is for R&D use only, not for drug, (IGF-1), and has been shown to enhance household, or other uses. Please consult the Material + + 3 Na /K -ATPase activity in a variety of cells. In Safety Data Sheet for information regarding hazards addition, SGK2 mimics the function of SGK1 and SGK3 + and safe handling practices. and participates in the regulation of renal epithelial Na 4 channel ENaC activity. Preparation instructions For maximum product recovery, after thawing, SGK2 is activated by phosphorylation in response to centrifuge the vial before removing the cap signals that stimulate PI3-kinase by a large number of 5 extracellular signals. -

Mtor- and SGK-Mediated Connexin 43 Expression Participates in Lipopolysaccharide-Stimulated Macrophage Migration Through the Inos/Src/FAK Axis
The Journal of Immunology mTOR- and SGK-Mediated Connexin 43 Expression Participates in Lipopolysaccharide-Stimulated Macrophage Migration through the iNOS/Src/FAK Axis Chen Shen,* Jin Hong Chen,* Youngyi Lee,† Md. Mehedi Hassan,‡ Su Jin Kim,* Eun Young Choi,x Seong-Tshool Hong,‡ Byung-Hyun Park,† and Ji Hyun Park* Connexin 43 (Cx43) deficiency was found to increase mortality in a mouse model of bacterial peritonitis, and Cx43 is upregulated in macrophages by LPS treatment. In this study, we characterized a novel signaling pathway for LPS-induced Cx43 expression in RAW264.7 cells and thioglycolate-elicited peritoneal macrophages (TGEMs). LPS alone or LPS-containing conditioned medium (CM) upregulated Cx43. Overexpression or silencing of Cx43 led to the enhancement or inhibition, respectively, of CM-induced TGEM migration. This response involved the inducible NO synthase (iNOS)/focal adhesion kinase (FAK)/Src pathways. Moreover, CM-induced migration was compromised in TGEMs from Cx43+/2 mice compared with TGEMs from Cx43+/+ littermates. Cx43 was upregulated by a serum/glucocorticoid-regulated kinase 1 (SGK) activator and downregulated, along with inhibition of CM- induced TGEM migration, by knockdown of the SGK gene or blockade of the SGK pathway. LPS-induced SGK activation was abrogated by Torin2, whereas LPS-induced Cx43 was downregulated by both Torin2 and rapamycin. Analysis of the effects of FK506 and methylprednisolone, common immunosuppressive agents following organ transplantation, suggested a link between these immunosuppressive drugs and impaired macrophage migration via the Cx43/iNOS/Src/FAK pathway. In a model of Escherichia coli infectious peritonitis, GSK650349-, an SGK inhibitor, or Torin2-treated mice showed less accumulation of F4/80+CD11b+ macrophages in the peritoneal cavity, with a delay in the elimination of bacteria. -

Sgk: an Old Enzyme Revisited
Sgk: an old enzyme revisited Nicolette Farman, … , Sheerazed Boulkroun, Nathalie Courtois-Coutry J Clin Invest. 2002;110(9):1233-1234. https://doi.org/10.1172/JCI17064. Commentary The elucidation of a key role for the serum- and glucocorticoid-regulated kinase (Sgk) in the regulation of the epithelial Na+ channel (ENaC) has led to substantial advances in the understanding of the signaling pathways involved in corticosteroid hormone action (1, 2). Indeed it is well established that the mineralocorticoid hormone aldosterone is a major regulator of body fluid homeostasis and blood pressure levels, mainly because of its capacity to modulate renal sodium reabsorption. However many aspects of this regulation pathway are not fully understood. Aldosterone binds to the mineralocorticoid receptor (MR), a member of the nuclear receptor superfamily which controls a network of genes in tight epithelia, ultimately leading to regulation of sodium entry into epithelial cells via the amiloride-sensitive ENaC (3). Most of the early steps of aldosterone action following MR-mediated transcriptional events are still incompletely elucidated. Differential hybridization studies have identified the serine/threonine kinase Sgk as a major transcriptional target of aldosterone. At least two other isoforms of Sgk, Sgk2 and Sgk3, have been identified after the initial characterization of Sgk (1), now referred to as Sgk1 (4). In this issue of the JCI, Wulff and colleagues (5) generated sgk1- deficient mice and examined the effects of dietary salt-restriction on renal water and electrolyte excretion. Sgk1 is a positive regulator of sodium transport Within one hour of aldosterone exposure, […] Find the latest version: https://jci.me/17064/pdf COMMENTARY See the related article beginning on page 1263. -
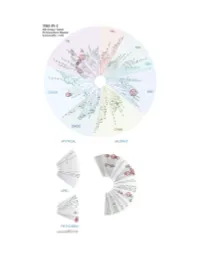
Profiling Data
Entrez Gene Percent Compound Compound Name DiscoveRx Gene Symbol Symbol Control Concentration (nM) THZ-P1-2 AAK1 AAK1 100 1000 THZ-P1-2 ABL1(E255K)-phosphorylated ABL1 21 1000 THZ-P1-2 ABL1(F317I)-nonphosphorylated ABL1 93 1000 THZ-P1-2 ABL1(F317I)-phosphorylated ABL1 100 1000 THZ-P1-2 ABL1(F317L)-nonphosphorylated ABL1 71 1000 THZ-P1-2 ABL1(F317L)-phosphorylated ABL1 44 1000 THZ-P1-2 ABL1(H396P)-nonphosphorylated ABL1 5.8 1000 THZ-P1-2 ABL1(H396P)-phosphorylated ABL1 6.6 1000 THZ-P1-2 ABL1(M351T)-phosphorylated ABL1 12 1000 THZ-P1-2 ABL1(Q252H)-nonphosphorylated ABL1 24 1000 THZ-P1-2 ABL1(Q252H)-phosphorylated ABL1 20 1000 THZ-P1-2 ABL1(T315I)-nonphosphorylated ABL1 93 1000 THZ-P1-2 ABL1(T315I)-phosphorylated ABL1 100 1000 THZ-P1-2 ABL1(Y253F)-phosphorylated ABL1 2.4 1000 THZ-P1-2 ABL1-nonphosphorylated ABL1 13 1000 THZ-P1-2 ABL1-phosphorylated ABL1 8.1 1000 THZ-P1-2 ABL2 ABL2 36 1000 THZ-P1-2 ACVR1 ACVR1 94 1000 THZ-P1-2 ACVR1B ACVR1B 100 1000 THZ-P1-2 ACVR2A ACVR2A 94 1000 THZ-P1-2 ACVR2B ACVR2B 91 1000 THZ-P1-2 ACVRL1 ACVRL1 90 1000 THZ-P1-2 ADCK3 CABC1 77 1000 THZ-P1-2 ADCK4 ADCK4 97 1000 THZ-P1-2 AKT1 AKT1 95 1000 THZ-P1-2 AKT2 AKT2 95 1000 THZ-P1-2 AKT3 AKT3 100 1000 THZ-P1-2 ALK ALK 92 1000 THZ-P1-2 ALK(C1156Y) ALK 93 1000 THZ-P1-2 ALK(L1196M) ALK 71 1000 THZ-P1-2 AMPK-alpha1 PRKAA1 93 1000 THZ-P1-2 AMPK-alpha2 PRKAA2 100 1000 THZ-P1-2 ANKK1 ANKK1 97 1000 THZ-P1-2 ARK5 NUAK1 83 1000 THZ-P1-2 ASK1 MAP3K5 100 1000 THZ-P1-2 ASK2 MAP3K6 95 1000 THZ-P1-2 AURKA AURKA 99 1000 THZ-P1-2 AURKB AURKB 100 1000 THZ-P1-2 AURKC AURKC 83 1000