Oximes: Novel Therapeutics with Anticancer and Anti-Inflammatory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

IRAK4 Gene Interleukin 1 Receptor Associated Kinase 4
IRAK4 gene interleukin 1 receptor associated kinase 4 Normal Function The IRAK4 gene provides instructions for making a protein that plays an important role in innate immunity, which is the body's early, nonspecific response to foreign invaders ( pathogens). The IRAK-4 protein is part of a signaling pathway that is involved in early recognition of pathogens and the initiation of inflammation to fight infection. In particular, the IRAK-4 protein relays signals from proteins called Toll-like receptors and IL-1 receptor-related proteins. As one of the first lines of defense against infection, Toll-like receptors recognize patterns that are common to many pathogens, rather than recognizing specific pathogens, and stimulate a quick immune response. The IL-1 receptor and related proteins recognize immune system proteins called cytokines that signal the need for an immune response. The resulting signaling pathway triggers inflammation, a nonspecific immune response that helps fight infection. Health Conditions Related to Genetic Changes IRAK-4 deficiency At least 20 mutations in the IRAK4 gene have been identified in people with IRAK-4 deficiency, an immune system disorder that leads to recurrent invasive bacterial infections. These gene mutations lead to an abnormally short, nonfunctional IRAK-4 protein or no protein at all. The loss of functional IRAK-4 protein blocks the initiation of inflammation in response to pathogens or cytokines that would normally help fight the infections. Because the early immune response is insufficient, bacterial -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: a Focus on the Present and an Outlook on the Future
biomolecules Review JAK-Inhibitors for the Treatment of Rheumatoid Arthritis: A Focus on the Present and an Outlook on the Future 1, 2, , 3 1,4 Jacopo Angelini y , Rossella Talotta * y , Rossana Roncato , Giulia Fornasier , Giorgia Barbiero 1, Lisa Dal Cin 1, Serena Brancati 1 and Francesco Scaglione 5 1 Postgraduate School of Clinical Pharmacology and Toxicology, University of Milan, 20133 Milan, Italy; [email protected] (J.A.); [email protected] (G.F.); [email protected] (G.B.); [email protected] (L.D.C.); [email protected] (S.B.) 2 Department of Clinical and Experimental Medicine, Rheumatology Unit, AOU “Gaetano Martino”, University of Messina, 98100 Messina, Italy 3 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico di Aviano (CRO), Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), Pordenone, 33081 Aviano, Italy; [email protected] 4 Pharmacy Unit, IRCCS-Burlo Garofolo di Trieste, 34137 Trieste, Italy 5 Head of Clinical Pharmacology and Toxicology Unit, Grande Ospedale Metropolitano Niguarda, Department of Oncology and Onco-Hematology, Director of Postgraduate School of Clinical Pharmacology and Toxicology, University of Milan, 20162 Milan, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-090-2111; Fax: +39-090-293-5162 Co-first authors. y Received: 16 May 2020; Accepted: 1 July 2020; Published: 5 July 2020 Abstract: Janus kinase inhibitors (JAKi) belong to a new class of oral targeted disease-modifying drugs which have recently revolutionized the therapeutic panorama of rheumatoid arthritis (RA) and other immune-mediated diseases, placing alongside or even replacing conventional and biological drugs. -
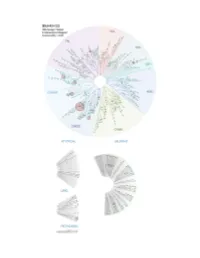
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

Glycogen Synthase Kinase-3 Is Activated in Neuronal Cells by G 12
The Journal of Neuroscience, August 15, 2002, 22(16):6863–6875 Glycogen Synthase Kinase-3 Is Activated in Neuronal Cells by G␣ ␣ 12 and G 13 by Rho-Independent and Rho-Dependent Mechanisms C. Laura Sayas, Jesu´ s Avila, and Francisco Wandosell Centro de Biologı´a Molecular “Severo Ochoa”, Consejo Superior de Investigaciones Cientı´ficas, Universidad Auto´ noma de Madrid, Cantoblanco, Madrid 28049, Spain ␣ ␣ ␣ ␣ Glycogen synthase kinase-3 (GSK-3) was generally considered tively active G 12 (G 12QL) and G 13 (G 13QL) in Neuro2a cells a constitutively active enzyme, only regulated by inhibition. induces upregulation of GSK-3 activity. Furthermore, overex- Here we describe that GSK-3 is activated by lysophosphatidic pression of constitutively active RhoA (RhoAV14) also activates ␣ acid (LPA) during neurite retraction in rat cerebellar granule GSK-3 However, the activation of GSK-3 by G 13 is blocked by neurons. GSK-3 activation correlates with an increase in GSK-3 coexpression with C3 transferase, whereas C3 does not block ␣ tyrosine phosphorylation. In addition, LPA induces a GSK-3- GSK-3 activation by G 12. Thus, we demonstrate that GSK-3 is ␣ ␣ mediated hyperphosphorylation of the microtubule-associated activated by both G 12 and G 13 in neuronal cells. However, ␣ protein tau. Inhibition of GSK-3 by lithium partially blocks neu- GSK-3 activation by G 13 is Rho-mediated, whereas GSK-3 ␣ rite retraction, indicating that GSK-3 activation is important but activation by G 12 is Rho-independent. The results presented not essential for the neurite retraction progress. GSK-3 activa- here imply the existence of a previously unknown mechanism of ␣ tion by LPA in cerebellar granule neurons is neither downstream GSK-3 activation by G 12/13 subunits. -

Supplementary Materials
Supplementary Suppl. Figure 1: MAPK signalling pathway of A: NCI-H2502, B: NCI-H2452, C: MSTO-211H and D: MRC-5. Suppl. Figure 2: Cell cycle pathway of A: NCI-H2502, B: NCI-H2452, C: MSTO-211H and D: MRC- 5. Suppl. Figure 3: Cancer pathways of A: NCI-H2502, B: NCI-H2452, C: MSTO-211H and D: MRC-5. Suppl. Figure 4: Phosphorylation level of A: ARAF, B: EPHA1, C: EPHA2, D: EPHA7 in all cell lines. For each cell line, phosphorylation levels are depicted before (Medium) and after cisplatin treatment (Cis). Suppl. Figure 5: Phosphorylation Level of A: KIT, B: PTPN11, C: PIK3R1, D: PTPN6 in all cell lines. For each cell line, phosphorylation levels are depicted before (Medium) and after cisplatin treatment (Cis). Suppl. Figure 6: Phosphorylation Level of A: KDR, B: EFS, C: AKT1, D: PTK2B/FAK2 in all cell lines. For each cell line, phosphorylation levels are depicted before (Medium) and after cisplatin treatment (Cis). Suppl. Figure 7: Scoreplots and volcanoplots of PTK upstream kinase analysis: A: Scoreplot of PTK- Upstream kinase analysis for NCI-H2052 cells. B: Volcanoplot of PTK-Upstream kinase analysis for NCI-H2052 cells. C: Scoreplot of PTK-Upstream kinase analysis for NCI-H2452 cells. D: Volcanoplot of PTK-Upstream kinase analysis for NCI-H2452 cells. E: Scoreplot of PTK-Upstream kinase analysis for MSTO-211H cells. F: Volcanoplot of PTK-Upstream kinase analysis for MSTO- 211H cells. G: Scoreplot of PTK-Upstream kinase analysis for MRC-5cells. H: Volcanoplot of PTK- Upstream kinase analysis for MRC-5 cells. Suppl. Figure 8: Scoreplots and volcanoplots of STK upstream kinase analysis: A: Scoreplot of STK- Upstream kinase analysis for NCI-H2052 cells. -

Targeting Myddosome Signaling in Waldenström’S
Author Manuscript Published OnlineFirst on August 20, 2018; DOI: 10.1158/1078-0432.CCR-17-3265 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. IRAK 1/4 Inhibition in Waldenström’s Ni, H. et al. Targeting Myddosome Signaling in Waldenström’s Macroglobulinemia with the Interleukin-1 Receptor- associated Kinase 1/4 Inhibitor R191 Haiwen Ni1,2,#, Fazal Shirazi2,#, Veerabhadran Baladandayuthapani3, Heather Lin3, Isere Kuiatse2, Hua Wang2, Richard J. Jones2, Zuzana Berkova2, Yasumichi Hitoshi4, Stephen M. Ansell5, Steven P. Treon6, Sheeba K. Thomas2, Hans C. Lee2, Zhiqiang Wang2, R. Eric Davis2, and Robert Z. Orlowski2,7,* 1Department of Hematology, The Affiliated Hospital of Nanjing University of Traditional Chinese Medicine, Nanjing, JangSu, China; 2Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, Houston, TX; 3Department of Biostatistics, The University of Texas MD Anderson Cancer Center, Houston, TX; 4Rigel, South San Francisco, CA; The 5Division of Hematology, Mayo Clinic, Rochester, MN; The 6Dana Farber Cancer Institute, Harvard Medical School, Boston, MA. 7Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX #Indicates that these authors contributed equally. Address correspondence to: Dr. Robert Z. Orlowski, The University of Texas MD Anderson Cancer Center, Department of Lymphoma and Myeloma, 1515 Holcombe Blvd., Unit 429, Houston, TX 77030-4009, E-mail: [email protected], Telephone 713-794-3234, Fax 713- 563-5067 Page 1 Downloaded from clincancerres.aacrjournals.org on September 24, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on August 20, 2018; DOI: 10.1158/1078-0432.CCR-17-3265 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Carna Newsletter 2021.05.26
画像:ファイル>配置>リンクから画像を選択>画像を選択して右クリック>画像を最前面に配置>オブジェクト>テキストの回り込み>作成 Vol.10 Carna Newsletter 2021.05.26 Targeted Degradation of Non-catalytic Kinases New Drug Discovery Options The announcement that Kymera Therapeutics, a TLR/IL-1R signaling in several cell types, which company pioneering targeted protein degradation, indicates that the IRAK4 scaffolding function is entered into a strategic collaboration with Sanofi important in some cases2)3). to develop and commercialize first-in-class protein degrader therapies targeting IRAK4 in patients with immune-inflammatory diseases highlights the TLR growing interest in clinical applications of small IL-1R molecule mediated kinase degradation. Kymera received $150 million in cash up front and may cytosol 8 8 potentially receive at least $2 billion in this key D y strategic partnership, including sales milestones M 4 4 K K and royalty payments. A A R R I I 2 P 1 K Most of the protein degraders currently under K A A R I R development are heterobifunctional molecules I which contain one moiety that binds a desired target protein and another that binds an E3 ligase, P joined by a linker. Protein degrader-induced proximity results in ubiquitination of the target Fig.1. TLR and IL-1R Signaling in the Myddosome followed by its degradation by the proteasome. This transformative new modality is expected to open a new chapter in drug discovery targeting 【Allosteric regulatory function】 kinases for which the development of clinical Aside from scaffolding functions, allosteric inhibitors has been difficult. One such example is regulation is another non-catalytic function that IRAK4, which has a non-catalytic function activates binding partners by inducing a independent of kinase activity in addition to a conformational change. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

Supplementary Table 1. in Vitro Side Effect Profiling Study for LDN/OSU-0212320. Neurotransmitter Related Steroids
Supplementary Table 1. In vitro side effect profiling study for LDN/OSU-0212320. Percent Inhibition Receptor 10 µM Neurotransmitter Related Adenosine, Non-selective 7.29% Adrenergic, Alpha 1, Non-selective 24.98% Adrenergic, Alpha 2, Non-selective 27.18% Adrenergic, Beta, Non-selective -20.94% Dopamine Transporter 8.69% Dopamine, D1 (h) 8.48% Dopamine, D2s (h) 4.06% GABA A, Agonist Site -16.15% GABA A, BDZ, alpha 1 site 12.73% GABA-B 13.60% Glutamate, AMPA Site (Ionotropic) 12.06% Glutamate, Kainate Site (Ionotropic) -1.03% Glutamate, NMDA Agonist Site (Ionotropic) 0.12% Glutamate, NMDA, Glycine (Stry-insens Site) 9.84% (Ionotropic) Glycine, Strychnine-sensitive 0.99% Histamine, H1 -5.54% Histamine, H2 16.54% Histamine, H3 4.80% Melatonin, Non-selective -5.54% Muscarinic, M1 (hr) -1.88% Muscarinic, M2 (h) 0.82% Muscarinic, Non-selective, Central 29.04% Muscarinic, Non-selective, Peripheral 0.29% Nicotinic, Neuronal (-BnTx insensitive) 7.85% Norepinephrine Transporter 2.87% Opioid, Non-selective -0.09% Opioid, Orphanin, ORL1 (h) 11.55% Serotonin Transporter -3.02% Serotonin, Non-selective 26.33% Sigma, Non-Selective 10.19% Steroids Estrogen 11.16% 1 Percent Inhibition Receptor 10 µM Testosterone (cytosolic) (h) 12.50% Ion Channels Calcium Channel, Type L (Dihydropyridine Site) 43.18% Calcium Channel, Type N 4.15% Potassium Channel, ATP-Sensitive -4.05% Potassium Channel, Ca2+ Act., VI 17.80% Potassium Channel, I(Kr) (hERG) (h) -6.44% Sodium, Site 2 -0.39% Second Messengers Nitric Oxide, NOS (Neuronal-Binding) -17.09% Prostaglandins Leukotriene, -

Protein Kinase C As a Paradigm Alexandra C
Biochem. J. (2003) 370, 361–371 (Printed in Great Britain) 361 REVIEW ARTICLE Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm Alexandra C. NEWTON1 Department of Pharmacology, University of California at San Diego, La Jolla, CA 92093-0640, U.S.A. Phosphorylation plays a central role in regulating the activation down-regulation of PKC as a paradigm for how these sites and signalling lifetime of protein kinases A, B (also known as control the function of the ABC kinases. Akt) and C. These kinases share three conserved phosphorylation motifs: the activation loop segment, the turn motif and the Key words: phosphoinositide 3-kinase, phosphoinositide-depen- hydrophobic motif. This review focuses on how phosphorylation dent kinase-1 (PDK-1), phosphorylation motif, protein kinase A at each of these sites regulates the maturation, signalling and (PKA), protein kinase B (PKB)\Akt, protein kinase C (PKC). INTRODUCTION by phosphorylation as a paradigm for control of ABC kinase function by phosphorylation. Almost half a century ago Krebs, Graves and Fischer reported b that the first discovered protein kinase, phosphorylase kinase, ARCHITECTURE OF ABC KINASES was, itself, activated by phosphorylation [1]. Two decades later, Fischer and colleagues showed that the kinase that phosphory- Protein kinases A, B\Akt and C have in common a conserved lates phosphorylase kinase, later named protein kinase A (PKA), kinase core whose function is regulated allosterically by a was also phosphorylated [2]. Reversible control by phosphory- corresponding regulatory moiety (Figure 1). In the case of PKA, lation\dephosphorylation is now firmly established as a fun- the regulatory moiety is a separate polypeptide, whereas the damental mechanism for regulating the function of most of the regulatory determinants are on the same polypeptide as the 500 or so members of the mammalian protein kinase superfamily. -

A Closer Look at JAK/STAT Signaling Pathway Emira Bousoik Chapman University
Chapman University Chapman University Digital Commons Pharmacy Faculty Articles and Research School of Pharmacy 7-31-2018 “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway Emira Bousoik Chapman University Hamidreza Montazeri Aliabadi Chapman University, [email protected] Follow this and additional works at: https://digitalcommons.chapman.edu/pharmacy_articles Part of the Amino Acids, Peptides, and Proteins Commons, Cancer Biology Commons, Cell Anatomy Commons, Cell Biology Commons, Enzymes and Coenzymes Commons, Oncology Commons, and the Other Pharmacy and Pharmaceutical Sciences Commons Recommended Citation Bousoik E, Montazeri Aliabadi H. “Do we know jack” about JAK? A closer look at JAK/STAT signaling pathway. Front. Oncol. 2018;8:287. doi: 10.3389/fonc.2018.00287 This Article is brought to you for free and open access by the School of Pharmacy at Chapman University Digital Commons. It has been accepted for inclusion in Pharmacy Faculty Articles and Research by an authorized administrator of Chapman University Digital Commons. For more information, please contact [email protected]. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway Comments This article was originally published in Frontiers in Oncology, volume 8, in 2018. DOI: 10.3389/ fonc.2018.00287 Creative Commons License This work is licensed under a Creative Commons Attribution 4.0 License. Copyright The uthora s This article is available at Chapman University Digital Commons: https://digitalcommons.chapman.edu/pharmacy_articles/590