Different Evolutionary Processes Shaped the Mouse and Human Olfactory Receptor Gene Families Janet M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Database Tool the Systematic Annotation of the Three Main GPCR
Database, Vol. 2010, Article ID baq018, doi:10.1093/database/baq018 ............................................................................................................................................................................................................................................................................................. Database tool The systematic annotation of the three main Downloaded from https://academic.oup.com/database/article-abstract/doi/10.1093/database/baq018/406672 by guest on 15 January 2019 GPCR families in Reactome Bijay Jassal1, Steven Jupe1, Michael Caudy2, Ewan Birney1, Lincoln Stein2, Henning Hermjakob1 and Peter D’Eustachio3,* 1European Bioinformatics Institute, Hinxton, Cambridge, CB10 1SD, UK, 2Ontario Institute for Cancer Research, Toronto, ON M5G 0A3, Canada and 3New York University School of Medicine, New York, NY 10016, USA *Corresponding author: Tel: +212 263 5779; Fax: +212 263 8166; Email: [email protected] Submitted 14 April 2010; Revised 14 June 2010; Accepted 13 July 2010 ............................................................................................................................................................................................................................................................................................. Reactome is an open-source, freely available database of human biological pathways and processes. A major goal of our work is to provide an integrated view of cellular signalling processes that spans from ligand–receptor -

Genomic Selection Signatures in Sheep from the Western Pyrenees Otsanda Ruiz-Larrañaga, Jorge Langa, Fernando Rendo, Carmen Manzano, Mikel Iriondo, Andone Estonba
Genomic selection signatures in sheep from the Western Pyrenees Otsanda Ruiz-Larrañaga, Jorge Langa, Fernando Rendo, Carmen Manzano, Mikel Iriondo, Andone Estonba To cite this version: Otsanda Ruiz-Larrañaga, Jorge Langa, Fernando Rendo, Carmen Manzano, Mikel Iriondo, et al.. Genomic selection signatures in sheep from the Western Pyrenees. Genetics Selection Evolution, BioMed Central, 2018, 50 (1), pp.9. 10.1186/s12711-018-0378-x. hal-02405217 HAL Id: hal-02405217 https://hal.archives-ouvertes.fr/hal-02405217 Submitted on 11 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Ruiz-Larrañaga et al. Genet Sel Evol (2018) 50:9 https://doi.org/10.1186/s12711-018-0378-x Genetics Selection Evolution RESEARCH ARTICLE Open Access Genomic selection signatures in sheep from the Western Pyrenees Otsanda Ruiz‑Larrañaga1* , Jorge Langa1, Fernando Rendo2, Carmen Manzano1, Mikel Iriondo1 and Andone Estonba1 Abstract Background: The current large spectrum of sheep phenotypic diversity -

Evaluating Recovery Potential of the Northern White Rhinoceros from Cryopreserved Somatic Cells
Downloaded from genome.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Research Evaluating recovery potential of the northern white rhinoceros from cryopreserved somatic cells Tate Tunstall,1 Richard Kock,2 Jiri Vahala,3 Mark Diekhans,4 Ian Fiddes,4 Joel Armstrong,4 Benedict Paten,4 Oliver A. Ryder,1,5 and Cynthia C. Steiner1,5 1San Diego Zoo Institute for Conservation Research, Escondido, California 92027, USA; 2Royal Veterinary College, University of London, London NW1 0TU, United Kingdom; 3Dvur Krlov Zoo, Dvr Krlov nad Labem 544 01, Czech Republic; 4Jack Baskin School of Engineering, University California Santa Cruz, Santa Cruz, California 95064, USA The critically endangered northern white rhinoceros is believed to be extinct in the wild, with the recent death of the last male leaving only two remaining individuals in captivity. Its extinction would appear inevitable, but the development of advanced cell and reproductive technologies such as cloning by nuclear transfer and the artificial production of gametes via stem cells differentiation offer a second chance for its survival. In this work, we analyzed genome-wide levels of genetic diversity, inbreeding, population history, and demography of the white rhinoceros sequenced from cryopreserved somatic cells, with the goal of informing how genetically valuable individuals could be used in future efforts toward the genetic res- cue of the northern white rhinoceros. We present the first sequenced genomes of the northern white rhinoceros, which show relatively high levels of heterozygosity and an average genetic divergence of 0.1% compared with the southern sub- species. The two white rhinoceros subspecies appear to be closely related, with low genetic admixture and a divergent time <80,000 yr ago. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -
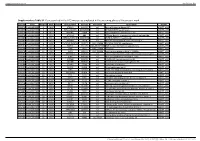
Supplementary Table S1. Genes Printed in the HC5 Microarray Employed in the Screening Phase of the Present Work
Supplementary material Ann Rheum Dis Supplementary Table S1. Genes printed in the HC5 microarray employed in the screening phase of the present work. CloneID Plate Position Well Length GeneSymbol GeneID Accession Description Vector 692672 HsxXG013989 2 B01 STK32A 202374 null serine/threonine kinase 32A pANT7_cGST 692675 HsxXG013989 3 C01 RPS10-NUDT3 100529239 null RPS10-NUDT3 readthrough pANT7_cGST 692678 HsxXG013989 4 D01 SPATA6L 55064 null spermatogenesis associated 6-like pANT7_cGST 692679 HsxXG013989 5 E01 ATP1A4 480 null ATPase, Na+/K+ transporting, alpha 4 polypeptide pANT7_cGST 692689 HsxXG013989 6 F01 ZNF816-ZNF321P 100529240 null ZNF816-ZNF321P readthrough pANT7_cGST 692691 HsxXG013989 7 G01 NKAIN1 79570 null Na+/K+ transporting ATPase interacting 1 pANT7_cGST 693155 HsxXG013989 8 H01 TNFSF12-TNFSF13 407977 NM_172089 TNFSF12-TNFSF13 readthrough pANT7_cGST 693161 HsxXG013989 9 A02 RAB12 201475 NM_001025300 RAB12, member RAS oncogene family pANT7_cGST 693169 HsxXG013989 10 B02 SYN1 6853 NM_133499 synapsin I pANT7_cGST 693176 HsxXG013989 11 C02 GJD3 125111 NM_152219 gap junction protein, delta 3, 31.9kDa pANT7_cGST 693181 HsxXG013989 12 D02 CHCHD10 400916 null coiled-coil-helix-coiled-coil-helix domain containing 10 pANT7_cGST 693184 HsxXG013989 13 E02 IDNK 414328 null idnK, gluconokinase homolog (E. coli) pANT7_cGST 693187 HsxXG013989 14 F02 LYPD6B 130576 null LY6/PLAUR domain containing 6B pANT7_cGST 693189 HsxXG013989 15 G02 C8orf86 389649 null chromosome 8 open reading frame 86 pANT7_cGST 693194 HsxXG013989 16 H02 CENPQ 55166 -

Genetic Characterization of Greek Population Isolates Reveals Strong Genetic Drift at Missense and Trait-Associated Variants
ARTICLE Received 22 Apr 2014 | Accepted 22 Sep 2014 | Published 6 Nov 2014 DOI: 10.1038/ncomms6345 OPEN Genetic characterization of Greek population isolates reveals strong genetic drift at missense and trait-associated variants Kalliope Panoutsopoulou1,*, Konstantinos Hatzikotoulas1,*, Dionysia Kiara Xifara2,3, Vincenza Colonna4, Aliki-Eleni Farmaki5, Graham R.S. Ritchie1,6, Lorraine Southam1,2, Arthur Gilly1, Ioanna Tachmazidou1, Segun Fatumo1,7,8, Angela Matchan1, Nigel W. Rayner1,2,9, Ioanna Ntalla5,10, Massimo Mezzavilla1,11, Yuan Chen1, Chrysoula Kiagiadaki12, Eleni Zengini13,14, Vasiliki Mamakou13,15, Antonis Athanasiadis16, Margarita Giannakopoulou17, Vassiliki-Eirini Kariakli5, Rebecca N. Nsubuga18, Alex Karabarinde18, Manjinder Sandhu1,8, Gil McVean2, Chris Tyler-Smith1, Emmanouil Tsafantakis12, Maria Karaleftheri16, Yali Xue1, George Dedoussis5 & Eleftheria Zeggini1 Isolated populations are emerging as a powerful study design in the search for low-frequency and rare variant associations with complex phenotypes. Here we genotype 2,296 samples from two isolated Greek populations, the Pomak villages (HELIC-Pomak) in the North of Greece and the Mylopotamos villages (HELIC-MANOLIS) in Crete. We compare their genomic characteristics to the general Greek population and establish them as genetic isolates. In the MANOLIS cohort, we observe an enrichment of missense variants among the variants that have drifted up in frequency by more than fivefold. In the Pomak cohort, we find novel associations at variants on chr11p15.4 showing large allele frequency increases (from 0.2% in the general Greek population to 4.6% in the isolate) with haematological traits, for example, with mean corpuscular volume (rs7116019, P ¼ 2.3 Â 10 À 26). We replicate this association in a second set of Pomak samples (combined P ¼ 2.0 Â 10 À 36). -

Human Olfactory Receptors
Human Olfactory Receptors: A journey from cell engineering for efficient in vitro functional assays to effective antagonists in human sensory assay Huysseune Sandra, Philippeau Magali, Moreau Cédric, Veithen Alex, Chatelain Pierre, Quesnel Yannick. ChemCom S.A., Route de Lennik 802, 1070 Anderlecht, Belgium. Cell lines Introduction: ChemCom's Entry ORs Literature data HEK293T HANA3 proprietary Many odorant compounds are perceived as unpleasant. They can be present in different contexts such as body-, home-, factory-, material-, or fabric-emitted 1 OR56A1 Adipietro et al., 2012 odors, so that humans are daily exposed to this olfactory pollution. In addition, odorant compounds can also taint food or beverages. Malodor and off-note 2 OR10J5 Saito et al., 2009 counteraction is a daily challenge for many different industries. 3 ORX116 - The first step of the odor perception corresponds to the interaction of olfactory receptors (ORs) with odorant molecules. Therefore, a selective inhibition of the 4 ORX069 - ORs by weakly odorant or odorless antagonists represents an innovative solution to malodor issues. The identification of such odor blockers requires an 5 OR10H5 - efficient technological platform to first fish out the receptors that interact with a malodor of interest and second, to screen libraries of potential antagonists of 6 OR8B3 Mainland et al., 2013 7 OR6P1 Mainland et al., 2013 these ORs. 8 ORX074B - 9 ORX126 - Materials and Methods: 10 ORX189 - 11 ORX081 - In vitro functional assay 12 ORX213 - Dilution-response analysis were performed in HEK293T, HANA3 and HEK293T-hRTP1S/hRTP2 cells using the CRE-luciferase reporter assay system. Briefly, each 13 OR1D2 Spehr et al., 2003 cell plated one day before was transfected with deorphanized ORs (identified at Chemcom S.A.) or empty vector plasmids using TransIT®-LT1 (Mirus) 14 OR7C1 Mainland et al., 2013 according to the manufacturer’s protocol. -

Contents 1 Northern White Rhinoceros Pedigree
Contents 1 Northern white rhinoceros pedigree 1 2 Genetic Divergence 2 3 Shared SNP Polymorphism 2 4 Admixture and PCA 4 5 Mitochondrial Tree 5 6 Demographic Inference Using @a@i 7 7 Inbreeding 8 8 Selection 9 9 Identification of the X chromosome 14 1 Northern white rhinoceros pedigree Figure 1: NWR pedigree highlighting individuals sequenced in this study (in blue box) presumably unrelated, with name, studbook number, ID number, and ploidy number 1 Dinka Lucy Angalifu Nola Nadi SB #74 SB #28 SB #348 SB #374 SB #376 KB6571- KB3731- KB9947- KB 8175 KB 5764 1845 618 3803 FZ 2859 FZ 1329 2n = 82 2n = 82 2n = 82 2n = 82 2n = 82 Nasima Saut SB #351 SB #373 KB8174- KB9939- 2872 3799 2n = 82 2n = 82 Nabire Suni SB #789 SB #630 KB8172- KB5765- 2864 2863 2n = 81 2n = 82 2 Genetic Divergence Pair-wise genetic divergence was estimated between all pairs of individuals using sites callable among all individuals, and defined as (2*homs+hets)/(2*callable fraction of genome), as defined in (Prado-Martinez et al., 2013). Divergence values for all individuals are shown in Supplementary Material Table 1. Calcu- lations were performed on the full set of 9.4 million SNPs. 3 Shared SNP Polymorphism In order to calculate shared polymorphism between the NWR and SWR, we took the average polymorphism of all possible combinations of the nine NWR 2 Table 1: Pair-wise genetic divergence for all rhinoceroses included in this study SB 28 SB 377 SB 376 SB 372 SB 156 SB 74 SB 24 SB 147 SB 351 SB 374 SB 373 SB 348 SB 34 SB 28 0 0 0 0 0 0 0 0 0 0 0 0 0 SB 377 0.0020231648 -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

UV-Irradiation- and Inflammation-Induced Skin Barrier Dysfunction Is Associated with the Expression of Olfactory Receptor Genes
International Journal of Molecular Sciences Article UV-Irradiation- and Inflammation-Induced Skin Barrier Dysfunction Is Associated with the Expression of Olfactory Receptor Genes in Human Keratinocytes Wesuk Kang, Bomin Son, Soyoon Park, Dabin Choi and Taesun Park * Department of Food and Nutrition, BK21 FOUR, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 120-749, Korea; [email protected] (W.K.); [email protected] (B.S.); [email protected] (S.P.); [email protected] (D.C.) * Correspondence: [email protected]; Tel.: +82-2-2123-3123; Fax: +82-2-365-3118 Abstract: Olfactory receptors (ORs) have diverse physiological roles in various cell types, beyond their function as odorant sensors in the olfactory epithelium. These previous findings have suggested that ORs could be diagnostic markers and promising therapeutic targets in several pathological conditions. In the current study, we sought to characterize the changes in the expression of ORs in the HaCaT human keratinocytes cell line exposed to ultraviolet (UV) light or inflammation, well-recognized stimulus for skin barrier disruption. We confirmed that major olfactory signaling components, including ORs, GNAL, Ric8b, and adenylate cyclase type 3, are highly expressed in HaCaT cells. We have also demonstrated that the 12 ectopic ORs detectable in HaCaT cells are more highly expressed in UV-irradiated or inflamed conditions than in normal conditions. We further assessed the specific OR-mediated biological responses of HaCaT cells in the presence of known odorant ligands of ORs and observed that specific ligand-activated ORs downregulate skin barrier Citation: Kang, W.; Son, B.; Park, S.; genes in HaCaT cells. This study shows the potential of OR as a marker for skin barrier abnormalities. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e .