Prion Diseases in Knock-In Mice Carrying Single Prp Codon Substitutions Associated with Human Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pnas11148toc 3..7
December 2, 2014 u vol. 111 u no. 48 u 16975–17336 Cover image: Pictured is a vermillion sea star, Mediaster aequalis,inPorlierPass,British Columbia, that shows signs of sea-star wasting disease. As of June 2014, the disease had affected 20 species of sea stars from Alaska to Baja California, but its cause is unknown. Ian Hewson et al. surveyed sea-star populations and conducted laboratory infection studies. They found that sea-star wasting disease is likely caused by a virus and identified a densovirus as a potential infectious agent. See the article by Hewson et al. on pages 17278–17283. Image courtesy of Peter Luckham (www.divemaster.ca). From the Cover 17278 Potential cause of sea-star wasting disease 17075 Human ability to evaluate probabilities 17122 X-ray free crystallography 17140 DNA binding site recognition by regulatory proteins 17182 Ebola antibodies’ modes of action Contents COMMENTARIES 16982 Going viral and the fatal vulnerability of neurons from immunity, not from infection THIS WEEK IN PNAS Lawrence Steinman See companion article on page 16053 in issue 45 of volume 111 16975 In This Issue 16984 Fuzzy universality of probability judgment Valerie F. Reyna and Charles J. Brainerd See companion article on page 17075 INNER WORKINGS—An over-the-shoulder look at scientists at work 16986 Expanding the femtosecond crystallography toolkit Sol M. Gruner 16977 Inner Workings: Freeing the dinos within See companion article on page 17122 Stephen Ornes CORE CONCEPTS—A brief introduction to emerging topics in science PNAS PLUS 16978 Core Concept: Synthetic biology—change, accelerated 16988 Significance Statements Danielle Venton Brief statements written by the authors about the significance of their papers. -

Vaccination with Prion Peptide-Displaying Polyomavirus-Like Particles Prolongs Incubation Time in Scrapie-Infected Mice
viruses Article Vaccination with Prion Peptide-Displaying Polyomavirus-Like Particles Prolongs Incubation Time in Scrapie-Infected Mice Martin Eiden 1,* , Alma Gedvilaite 2 , Fabienne Leidel 1,3, Rainer G. Ulrich 1 and Martin H. Groschup 1 1 Institute of Novel and Emerging Infectious Diseases, Friedrich-Loeffler-Institut, Federal Research Institute for Animal Health, Südufer 10, 17493 Greifswald-Insel Riems, Germany; [email protected] (F.L.); rainer.ulrich@fli.de (R.G.U.); martin.groschup@fli.de (M.H.G.) 2 Life Sciences Center, Institute of Biotechnology, Vilnius University, Sauletekio˙ al. 7, LT-10257 Vilnius, Lithuania; [email protected] 3 Task Force Animal Diseases, Darmstadt Regional Administrative Council, Luisenplatz 2, 64283 Darmstadt, Germany * Correspondence: martin.eiden@fli.de Abstract: Prion diseases like scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle or Creutzfeldt–Jakob disease (CJD) in humans are fatal neurodegenerative diseases characterized by the conformational conversion of the normal, mainly α-helical cellular prion protein (PrPC) into the abnormal β-sheet rich infectious isoform PrPSc. Various therapeutic or prophylactic approaches have been conducted, but no approved therapeutic treatment is available so far. Immunisation against prions is hampered by the self-tolerance to PrPC in mammalian species. One strategy to avoid this tolerance is presenting PrP variants in virus-like particles (VLPs). Therefore, we vaccinated C57/BL6 mice with nine prion peptide variants presented by hamster polyomavirus capsid protein VP1/VP2-derived VLPs. Mice were subsequently challenged intraperitoneally with the murine RML Citation: Eiden, M.; Gedvilaite, A.; prion strain. Importantly, one group exhibited significantly increased mean survival time of 240 days Leidel, F.; Ulrich, R.G.; Groschup, M.H. -

Hria Medical Foundation 2012 Review
20․12 DI VISION REVIE W Where Science and Philanthropy Converge IN THIS ISSUE: About Us and Our Services / 2 Funding Opportunities Jeffress Trust /8 Hood Foundation /10 Noonan Memorial Research Fund /11 Klarman Family Foundation /12 King Trust /13 Thome Foundation /14 Smith Family Foundation /16 Davis Foundation /17 Lymphatic Research Foundation /18 Scientific Review Committees /19 Gene Discovery in Anorexia Nervosa / 6 The Medical Foundation, a division of HRiA About Us Since 1957, foundations, bank trusts and individuals have engaged us to create and manage customized biomedical research grant programs that accelerate the pace of scientific discoveries. As evidenced by the more than 145,000 visits to our website this year alone, our funding announcements reach thousands of potential applicants for every grant cycle. And, by building a distinguished Scientific Review Committee for each program, we ensure critical and unbiased selection of the best minds in science. In 2012, we were privileged to work with foundations and bank trust departments whose grant programs distributed more than $18 million to investigators and physician-scientists across the United States and worldwide. Sally E. McNagny, M.D., M.P.H., F.A.C.P., Vice President Since 2001, Dr. McNagny has served as Vice President and head of HRiA’s Medical Foundation division where she leads biomedical research grantmaking and life sciences consulting. Dr. McNagny also serves on the faculty at Harvard Medical School and is a Fellow of the American College of Physicians. She holds a B.S. in Biology from Stanford University, an M.D. from Harvard Medical School, an M.P.H. -

Zinc and Copper Ions Differentially Regulate Prion-Like Phase
viruses Article Zinc and Copper Ions Differentially Regulate Prion-Like Phase Separation Dynamics of Pan-Virus Nucleocapsid Biomolecular Condensates Anne Monette 1,* and Andrew J. Mouland 1,2,* 1 Lady Davis Institute at the Jewish General Hospital, Montréal, QC H3T 1E2, Canada 2 Department of Medicine, McGill University, Montréal, QC H4A 3J1, Canada * Correspondence: [email protected] (A.M.); [email protected] (A.J.M.) Received: 2 September 2020; Accepted: 12 October 2020; Published: 18 October 2020 Abstract: Liquid-liquid phase separation (LLPS) is a rapidly growing research focus due to numerous demonstrations that many cellular proteins phase-separate to form biomolecular condensates (BMCs) that nucleate membraneless organelles (MLOs). A growing repertoire of mechanisms supporting BMC formation, composition, dynamics, and functions are becoming elucidated. BMCs are now appreciated as required for several steps of gene regulation, while their deregulation promotes pathological aggregates, such as stress granules (SGs) and insoluble irreversible plaques that are hallmarks of neurodegenerative diseases. Treatment of BMC-related diseases will greatly benefit from identification of therapeutics preventing pathological aggregates while sparing BMCs required for cellular functions. Numerous viruses that block SG assembly also utilize or engineer BMCs for their replication. While BMC formation first depends on prion-like disordered protein domains (PrLDs), metal ion-controlled RNA-binding domains (RBDs) also orchestrate their formation. Virus replication and viral genomic RNA (vRNA) packaging dynamics involving nucleocapsid (NC) proteins and their orthologs rely on Zinc (Zn) availability, while virus morphology and infectivity are negatively influenced by excess Copper (Cu). While virus infections modify physiological metal homeostasis towards an increased copper to zinc ratio (Cu/Zn), how and why they do this remains elusive. -

Mitochondrial Dysfunction in Preclinical Genetic Prion Disease: a Target for 7 Preventive Treatment?
1HXURELRORJ\RI'LVHDVH ² Contents lists available at ScienceDirect Neurobiology of Disease journal homepage: www.elsevier.com/locate/ynbdi Mitochondrial dysfunction in preclinical genetic prion disease: A target for 7 preventive treatment? Guy Kellera,c,1, Orli Binyamina,c,1, Kati Frida,c, Ann Saadab,c, Ruth Gabizona,c,⁎ a Department of Neurology, The Agnes Ginges Center for Human Neurogenetics, Israel b Department of Genetics and Metabolic Diseases, The Monique and Jacques Roboh Department of Genetic Research, Hadassah-Hebrew University Medical Center, Israel c Medical School, The Hebrew University, Jerusalem, Israel ABSTRACT Mitochondrial malfunction is a common feature in advanced stages of neurodegenerative conditions, as is the case for the accumulation of aberrantly folded proteins, such as PrP in prion diseases. In this work, we investigated mitochondrial activity and expression of related factors vis a vis PrP accumulation at the subclinical stages of TgMHu2ME199K mice, modeling for genetic prion diseases. While these mice remain healthy until 5–6 months of age, they succumb to fatal disease at 12–14 months. We found that mitochondrial respiratory chain enzymatic activates and ATP/ROS production, were abnormally elevated in asymptomatic mice, concomitant with initial accumulation of disease related PrP. In parallel, the expression of Cytochrome c oxidase (COX) subunit IV isoform 1(Cox IV-1) was reduced and replaced by the activity of Cox IV isoform 2, which operates in oxidative neuronal conditions. At all stages of disease, Cox IV-1 was absent from cells accu- mulating disease related PrP, suggesting that PrP aggregates may directly compromise normal mitochondrial function. Administration of Nano-PSO, a brain targeted antioxidant, to TgMHu2ME199K mice, reversed functional and biochemical mitochondrial functions to normal conditions regardless of the presence of misfolded PrP. -

An Evolutionary Basis for Scrapie Disease : Identification of a Fish
72 Update TRENDS in Genetics Vol.19 No.2 February 2003 affect regulation of the human genome in a more global 11 Frith, M.C. et al. (2002) Statistical significance of clusters of motifs manner by creating S/MARs that form chromatin loops, represented by position specific scoring matrices in nucleotide and by shaping the sequence evolution of LCRs. sequences. Nucleic Acids Res. 30, 3214–3224 12 Wingender, E. et al. (2001) The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 29, 281–283 Acknowledgements 13 Purucker, M. et al. (1990) Structure and function of the enhancer 30 to Galina V. Glazko was supported by research grants from NIH (GM-20293) the human A gamma globin gene. Nucleic Acids Res. 18, 7407–7415 and NASA (NCC2-1057) awarded to Masatoshi Nei. We thank Nathan 14 Li, Q. et al. (1999) Locus control regions: coming of age at a decade plus. J. Bowen and Wolfgang J. Miller for discussions on the relationship Trends Genet. 15, 403–408 between TEs and S/MARs. 15 Hardison, R. et al. (1997) Locus control regions of mammalian beta- globin gene clusters: combining phylogenetic analyses and exper- References imental results to gain functional insights. Gene 205, 73–94 1 The Human Genome Sequencing Consortium, (2001) Initial sequen- 16 Strausberg, R. et al. (1999) The mammalian gene collection. Science cing and analysis of the human genome. Nature 409, 860–921 286, 455–457 2 Kidwell, M.G. and Lisch, D.R. (2001) Perspective: transposable 17 Bode, J. et al. (1996) Scaffold/matrix-attached regions: topological elements, parasitic DNA, and genome evolution. -

Structure & Symmetry
HAPPY HOLIDAYS ASBMB MEMBERS December 2008 Structure & Symmetry American Society for Biochemistry and Molecular Biology J\\PfliGifk\`ej n`k_*.#'''>=G$kX^^\[FI=Zcfe\j 8 $@C- 9 "@C- : ; >=G$kX^^\[Kil\FI=Zcfe\jXi\kiXej]\Zk\[`ekf?<B)0* Z\ccjXe[k_\kX^^\[gifk\`ejXi\m`jlXc`q\[[li`e^@C$- `e[lZ\[elZc\XikiXejcfZXk`feJK8K*#gXe\c8Xe[9 Xe[`e Ôcfgf[`XXe[jki\jjÔY\i]fidXk`fe8Zk`e#gXe\c:Xe[; % Kil\FI= Fi`>\e\jXclk\j >\efd\n`[\FI=Zcfe\j k_\>=Gg`fe\\ij ]fik_\`iEfY\c ]fikX^^\[gifk\`e\ogi\jj`fe Gi`q\XnXi[ ×:$k\id`eXckX^f]>=G ×J\hl\eZ\m\i`Ô\[Xe[^lXiXek\\[ ×<Xj`cpj_lkkc\[`ekf)'[\jk`eXk`fem\Zkfij ×KiXej]\Zk`fe$i\X[p1('l^gcXjd`[;E8 fi`^\e\%Zfd&fi] ORG-041-GFPTaggedAd_ASBMB_v7.indd 1 10/20/08 12:29:39 PM contents DECEMBER 2008 ON THE COVER: Captivated by the symmetry society news of molecular structure, Sung-Hou Kim has been a 2 From the Editor leader in revealing symmetry 3 President’s Message through his studies in crystallography and 5 Letters to the Editor structural genomics. 30 6 Washington Update 12 Retrospective: Anthony G. San Pietro FASEB releases new Breakthroughs in special interest Bioscience. 6 13 Science’s Role in Foreign Policy 14 ASBMB Round Table: Jim Wells and Mary Woolley 16 Keeping Women in Science 19 Grammar and Writing Tips 2009 meeting 20 The 2009 Fritz Lipmann Lectureship: Douglas C. Rees 21 The 2009 ASBMB Merck Award: John Kuriyan 22 The 2009 FASEB Excellence in Science Award: Susan Lindquist science focus 30 Sung-Hou Kim: Consummate Crystallographer departments 7 News from the Hill 10 Member Spotlight 23 Education and Training A leaky pipeline for women scientists. -

Susan Lee Lindquist (1949–2016)
In Memoriam Sue was a spectacular scientist who com- at the University of Chicago, where she bined a searing intellect with deep wis- would later join the faculty (1978) and rise Susan Lee Lindquist (1949–2016) dom, sagacious intuition, and limitless to full professor (1988). While at the Uni- creativity. These characteristics enabled versity of Chicago, Sue married Edward Sue to make connections across dispa- Buckbee and would have two wonderful 1, James Shorter * rate disciplines that nobody else could daughters, Alana and Nora. She also make. Her infectious esprit for scientific launched a remarkable and radical series Lindquist was a visionary and pio- discovery was combined with disarming of trailblazing discoveries. These contin- neer who transformed our under- warmth, positivity, openness, directness, ued when Sue moved her research pro- standing of how protein folding and generosity, which made her an inspi- gram to the Whitehead Institute for rational, nurturing, and indefatigable men- Biomedical Research at Massachusetts sculpts biology, evolution, and dis- tor. These synergistic traits empowered Institute of Technology (MIT), which is ease. She revealed several unantici- extraordinarily effective collaborations where I trained with Sue as a postdoctoral pated mechanisms by which protein between scientists from diverse back- fellow (2002–2007). Sue would spend the folding can buffer, release, and grounds and disciplines. Indeed, rest of her career at the Whitehead Insti- potentiate genetic variation in researchers from diverse backgrounds – tute as Director (2001–2004), Institute response to environmental stress, physicists, chemists, biochemists, biolo- Member (2001–2016), and Professor of thereby enabling the rapid evolution gists, mathematicians, and physicians – Biology at MIT (2001–2016). -

Calcineurin Determines Toxic Versus Beneficial Responses to Α-Synuclein
Calcineurin determines toxic versus beneficial responses to α-synuclein Gabriela Caraveoa,b, Pavan K. Aulucka,c,1, Luke Whitesella, Chee Yeun Chunga, Valeriya Barua,b, Eugene V. Mosharovd, Xiaohui Yane, Manu Ben-Johnyf, Martin Sosteg, Paola Picottig, Hanna Kime, Kim A. Caldwelle, Guy A. Caldwelle, David Sulzerd,h, David T. Yuef, and Susan Lindquista,b,2 aWhitehead Institute for Biomedical Research, Cambridge, MA 02142; bHoward Hughes Medical Institute, Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139; cDepartment of Pathology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114; Departments of dNeurology and hPsychiatry, Columbia University Medical Center, New York, NY 10032; eDepartment of Biological Sciences, The University of Alabama, Tuscaloosa, AL 35487; fDepartments of Biomedical Engineering and Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD 21205; and gDepartment of Biology, Institute of Biochemistry, Eidgenossische Technische Hochschule Zurich, Zurich CH-8093, Switzerland Contributed by Susan Lindquist, July 15, 2014 (sent for review May 7, 2014) + Calcineurin (CN) is a highly conserved Ca2 –calmodulin (CaM)- tigations, given their genetic tractability and the remarkable + + dependent phosphatase that senses Ca2 concentrations and trans- conservation of Ca2 -signaling pathways from yeast to humans duces that information into cellular responses. Ca2+ homeostasis (14, 15). Moreover, the expression of human α-syn in yeast leads is disrupted by α-synuclein (α-syn), a small lipid binding protein to cellular pathologies directly relevant to neurons and PD, in- whose misfolding and accumulation is a pathological hallmark of cluding nitrosative stress (16, 17), defects in vesicle trafficking several neurodegenerative diseases. We report that α-syn, from (18–20), and faulty mitochondrial function (21, 22). -
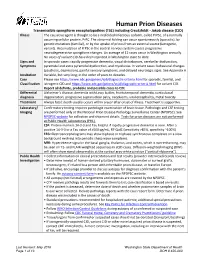
Prion Disease Reporting and Investigation Guideline
Human Prion Diseases Transmissible spongiform encephalopathies (TSE) including Creutzfeldt - Jakob disease (CJD) Illness The causative agent is thought to be a misfolded infectious isoform, called PrPSc, of a normally occurring cellular protein, PrPC. The abnormal folding can occur spontaneously (sporadic), by genetic mutations (familial), or by the uptake of prions from an external source (iatrogenic, variant). Accumulation of PrPSc in the central nervous system causes progressive neurodegenerative spongiform changes. An average of 12 cases occur in Washington annually. No cases of variant CJD have been reported in Washington state to date. Signs and In sporadic cases: rapidly progressive dementia, visual disturbances, cerebellar dysfunction, Symptoms pyramidal and extra pyramidal dysfunction, and myoclonus. In variant cases: behavioral changes (psychosis, depression), painful sensory symptoms, and delayed neurologic signs. See Appendix A Incubation Variable, but very long; in the order of years to decades. Case Please see https://www.cdc.gov/prions/cjd/diagnostic-criteria.html for sporadic, familial, and Classification iatrogenic CJD and https://www.cdc.gov/prions/vcjd/diagnostic-criteria.html for variant CJD. Report all definite, probable and possible cases to CDE Differential Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia, corticobasal diagnosis degeneration, progressive supranuclear palsy, neoplasms, viral encephalitis, metal toxicity Treatment Always fatal; death usually occurs within a year after onset of illness. Treatment is supportive. Laboratory/ Confirmatory testing requires pathologic examination of brain tissue. Pathologic and CSF testing Imaging are performed only at the National Prion Disease Pathology Surveillance Center (NPDPSC). See NPDPSC website for collection and shipment details. Tests for prion diseases are not performed at Public Health Laboratories (PHL). -

Lecture 1: INTRODUCTION in MEDICAL BIOLOGY. CELL STRUCTURE
Lecture 1: INTRODUCTION IN MEDICAL BIOLOGY. CELL STRUCTURE 1. Biology: The Science of Our Lives 2. Theories Contributing to Modern Biology: Cell Theory 3. Forms and Diversity of Life 4. Levels of Organization 5. Cell Structure Biology (from Greek βίος - life and λόγος - word, judgement) – is a branch of the natural sciences, and is the study of living organisms and their interactions with environment. The term was specially proposed by French naturalist Jean-Baptiste Pierre Antoine de Monet, Chevalier de Lamarck in 1802 Biology deals with every aspect of life in a living organism. Biology examines the structure, function, growth, origin, evolution, and distribution of living things. Modern biology is complex of sciences. Most biological sciences are specialized disciplines: Botany, Zoology, Protozoology, Microbiology, Virology, Molecular biology, Genetics, Embryology, Evolution theory, Ecology and so on. The history of biology traces the study of the living world from ancient to modern times. Although the concept of biology as a single coherent field arose in the 19th century, the biological sciences emerged from traditions of medicine and natural history reaching back to ancient Egyptian medicine and the works of Aristotle and Galen in the ancient Greco-Roman world, which were then further developed in the Middle Ages by Muslim physicians and scholars such as al-Jahiz, Avicenna, Avenzoar, Ibn al-Baitar and Ibn al-Nafis. Ancient Greek philosopher, Aristotle developed his Scala Naturae, or Ladder of Life, to explain his concept of the advancement -

The New Eppendorf Micro Centrifuge
The new Eppendorf Micro Centrifuge. • With 50% higher::capacity,: variable speed,. quieter operation, and .......... Brand ...: .......... Higher capacity..,plus. Safe and ru ggecJ. The new..1.8-place. Model 5415 The" Eppe~dorf 5415 Micro Micro Centrifuge g wes you Centrifuge is UL listed for ~.i.mportant operating advantages-- • . safety: It's sorugged th.atan with unique Eppendorf quality: : acci.den~tatlyiunbalanced 4oad ......... Enclosed rotor design reduces air.turbulence won't cause excessive vibration Versatile in use, and noise. Tubes are angled precisely at 45°.to 0rm0tof~ damage. M.odel.5415 has a variable-speed maximize pellet formation. For more information:: call " ' ~ : " motor that reaches a maximum 800~645-3050;. in NewYork., . of.14,000 rpm with anlRCF of 5164334.~7500.,:. Or write . 16,000 x g;. a 30-minute:timer; Brinkmann..Ins.truments, Inc., • and a momentary button for short Cantiague Road,. Westbury, spins. It accepts 1.5 mL,.500 t~L, NY 11590. (In Canada ................... 400 I~L, and 250 #L Eppendorf 416-~675W911; 50 Galaxy Blvd., •Microcentrifuge Tubes and Rexdale., Ont. M9W 4Y5) blood Collection microtubes, such as B-D Microtainer*Tubes. Specifications ......... Maximumspeed: l4,000i;pm " New rotor design. Maximum RCF ...... : 16.000 x g The enclosedrotor design Test-tube capacity ':: 1:.8 ...... reduces air turbulencefor Timerequired for " - " maximum speed i....10.sec. quieter operation:. And the new Timerequired to stop I~2 sec ........ - Quick, release feature; aitowsthe18-position :.Dimensions quick,reie.ase featu re lets.you rotor to be easily transported even.when ....... (L X W x H): 28 X 2! x 28.5 cm transport the: rotor with tubes--- Loaded .....