Mitochondrial Dysfunction in Preclinical Genetic Prion Disease: a Target for 7 Preventive Treatment?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vaccination with Prion Peptide-Displaying Polyomavirus-Like Particles Prolongs Incubation Time in Scrapie-Infected Mice
viruses Article Vaccination with Prion Peptide-Displaying Polyomavirus-Like Particles Prolongs Incubation Time in Scrapie-Infected Mice Martin Eiden 1,* , Alma Gedvilaite 2 , Fabienne Leidel 1,3, Rainer G. Ulrich 1 and Martin H. Groschup 1 1 Institute of Novel and Emerging Infectious Diseases, Friedrich-Loeffler-Institut, Federal Research Institute for Animal Health, Südufer 10, 17493 Greifswald-Insel Riems, Germany; [email protected] (F.L.); rainer.ulrich@fli.de (R.G.U.); martin.groschup@fli.de (M.H.G.) 2 Life Sciences Center, Institute of Biotechnology, Vilnius University, Sauletekio˙ al. 7, LT-10257 Vilnius, Lithuania; [email protected] 3 Task Force Animal Diseases, Darmstadt Regional Administrative Council, Luisenplatz 2, 64283 Darmstadt, Germany * Correspondence: martin.eiden@fli.de Abstract: Prion diseases like scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle or Creutzfeldt–Jakob disease (CJD) in humans are fatal neurodegenerative diseases characterized by the conformational conversion of the normal, mainly α-helical cellular prion protein (PrPC) into the abnormal β-sheet rich infectious isoform PrPSc. Various therapeutic or prophylactic approaches have been conducted, but no approved therapeutic treatment is available so far. Immunisation against prions is hampered by the self-tolerance to PrPC in mammalian species. One strategy to avoid this tolerance is presenting PrP variants in virus-like particles (VLPs). Therefore, we vaccinated C57/BL6 mice with nine prion peptide variants presented by hamster polyomavirus capsid protein VP1/VP2-derived VLPs. Mice were subsequently challenged intraperitoneally with the murine RML Citation: Eiden, M.; Gedvilaite, A.; prion strain. Importantly, one group exhibited significantly increased mean survival time of 240 days Leidel, F.; Ulrich, R.G.; Groschup, M.H. -

Zinc and Copper Ions Differentially Regulate Prion-Like Phase
viruses Article Zinc and Copper Ions Differentially Regulate Prion-Like Phase Separation Dynamics of Pan-Virus Nucleocapsid Biomolecular Condensates Anne Monette 1,* and Andrew J. Mouland 1,2,* 1 Lady Davis Institute at the Jewish General Hospital, Montréal, QC H3T 1E2, Canada 2 Department of Medicine, McGill University, Montréal, QC H4A 3J1, Canada * Correspondence: [email protected] (A.M.); [email protected] (A.J.M.) Received: 2 September 2020; Accepted: 12 October 2020; Published: 18 October 2020 Abstract: Liquid-liquid phase separation (LLPS) is a rapidly growing research focus due to numerous demonstrations that many cellular proteins phase-separate to form biomolecular condensates (BMCs) that nucleate membraneless organelles (MLOs). A growing repertoire of mechanisms supporting BMC formation, composition, dynamics, and functions are becoming elucidated. BMCs are now appreciated as required for several steps of gene regulation, while their deregulation promotes pathological aggregates, such as stress granules (SGs) and insoluble irreversible plaques that are hallmarks of neurodegenerative diseases. Treatment of BMC-related diseases will greatly benefit from identification of therapeutics preventing pathological aggregates while sparing BMCs required for cellular functions. Numerous viruses that block SG assembly also utilize or engineer BMCs for their replication. While BMC formation first depends on prion-like disordered protein domains (PrLDs), metal ion-controlled RNA-binding domains (RBDs) also orchestrate their formation. Virus replication and viral genomic RNA (vRNA) packaging dynamics involving nucleocapsid (NC) proteins and their orthologs rely on Zinc (Zn) availability, while virus morphology and infectivity are negatively influenced by excess Copper (Cu). While virus infections modify physiological metal homeostasis towards an increased copper to zinc ratio (Cu/Zn), how and why they do this remains elusive. -

Prion Diseases in Knock-In Mice Carrying Single Prp Codon Substitutions Associated with Human Diseases
Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases Walker S. Jacksona,b,c,1, Andrew W. Borkowskia,b,c, Nicki E. Watsona, Oliver D. Kingd, Henryk Faase, Alan Jasanoffe,f, and Susan Lindquista,b,c,2 aWhitehead Institute for Biomedical Research, Cambridge, MA 02142; bHoward Hughes Medical Institute, cDepartment of Biology, and fDepartments of Biological Engineering, Brain and Cognitive Sciences, and Nuclear Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139; dDepartment of Cell and Developmental Biology, University of Massachusetts Medical School, Worcester, MA 01655; and eFrances Bitter Magnet Laboratory, Cambridge, MA 02139 Contributed by Susan Lindquist, July 9, 2013 (sent for review March 7, 2013) In man, mutations in different regions of the prion protein (PrP) linked to it, provides an important general model for such are associated with infectious neurodegenerative diseases that investigations. have remarkably different clinical signs and neuropathological There are several types of human prion diseases, each begin- lesions. To explore the roots of this phenomenon, we created ning with pathologic processes in a different brain region and fi – a knock-in mouse model carrying the mutation associated with leading to distinct functional de cits: cognition [Creutzfeldt – – one of these diseases [Creutzfeldt–Jakob disease (CJD)] that was Jakob disease (CJD)], movement control (Gerstmann Sträussler Scheinker syndrome), or sleep and autonomic functions [fatal exactly analogous to a previous knock-in model of a different fl prion disease [fatal familial insomnia (FFI)]. Together with the familial insomnia (FFI)] (7). Prion diseases also af ict animals WT parent, this created an allelic series of three lines, each express- and include bovine spongiform encephalopathy (BSE) of cattle, scrapie of sheep and goats, and chronic wasting disease (CWD) ing the same protein with a single amino acid difference, and with of deer and elk (1). -

An Evolutionary Basis for Scrapie Disease : Identification of a Fish
72 Update TRENDS in Genetics Vol.19 No.2 February 2003 affect regulation of the human genome in a more global 11 Frith, M.C. et al. (2002) Statistical significance of clusters of motifs manner by creating S/MARs that form chromatin loops, represented by position specific scoring matrices in nucleotide and by shaping the sequence evolution of LCRs. sequences. Nucleic Acids Res. 30, 3214–3224 12 Wingender, E. et al. (2001) The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 29, 281–283 Acknowledgements 13 Purucker, M. et al. (1990) Structure and function of the enhancer 30 to Galina V. Glazko was supported by research grants from NIH (GM-20293) the human A gamma globin gene. Nucleic Acids Res. 18, 7407–7415 and NASA (NCC2-1057) awarded to Masatoshi Nei. We thank Nathan 14 Li, Q. et al. (1999) Locus control regions: coming of age at a decade plus. J. Bowen and Wolfgang J. Miller for discussions on the relationship Trends Genet. 15, 403–408 between TEs and S/MARs. 15 Hardison, R. et al. (1997) Locus control regions of mammalian beta- globin gene clusters: combining phylogenetic analyses and exper- References imental results to gain functional insights. Gene 205, 73–94 1 The Human Genome Sequencing Consortium, (2001) Initial sequen- 16 Strausberg, R. et al. (1999) The mammalian gene collection. Science cing and analysis of the human genome. Nature 409, 860–921 286, 455–457 2 Kidwell, M.G. and Lisch, D.R. (2001) Perspective: transposable 17 Bode, J. et al. (1996) Scaffold/matrix-attached regions: topological elements, parasitic DNA, and genome evolution. -
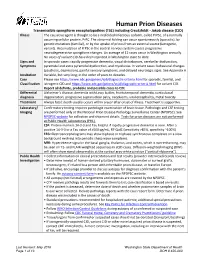
Prion Disease Reporting and Investigation Guideline
Human Prion Diseases Transmissible spongiform encephalopathies (TSE) including Creutzfeldt - Jakob disease (CJD) Illness The causative agent is thought to be a misfolded infectious isoform, called PrPSc, of a normally occurring cellular protein, PrPC. The abnormal folding can occur spontaneously (sporadic), by genetic mutations (familial), or by the uptake of prions from an external source (iatrogenic, variant). Accumulation of PrPSc in the central nervous system causes progressive neurodegenerative spongiform changes. An average of 12 cases occur in Washington annually. No cases of variant CJD have been reported in Washington state to date. Signs and In sporadic cases: rapidly progressive dementia, visual disturbances, cerebellar dysfunction, Symptoms pyramidal and extra pyramidal dysfunction, and myoclonus. In variant cases: behavioral changes (psychosis, depression), painful sensory symptoms, and delayed neurologic signs. See Appendix A Incubation Variable, but very long; in the order of years to decades. Case Please see https://www.cdc.gov/prions/cjd/diagnostic-criteria.html for sporadic, familial, and Classification iatrogenic CJD and https://www.cdc.gov/prions/vcjd/diagnostic-criteria.html for variant CJD. Report all definite, probable and possible cases to CDE Differential Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia, corticobasal diagnosis degeneration, progressive supranuclear palsy, neoplasms, viral encephalitis, metal toxicity Treatment Always fatal; death usually occurs within a year after onset of illness. Treatment is supportive. Laboratory/ Confirmatory testing requires pathologic examination of brain tissue. Pathologic and CSF testing Imaging are performed only at the National Prion Disease Pathology Surveillance Center (NPDPSC). See NPDPSC website for collection and shipment details. Tests for prion diseases are not performed at Public Health Laboratories (PHL). -

Lecture 1: INTRODUCTION in MEDICAL BIOLOGY. CELL STRUCTURE
Lecture 1: INTRODUCTION IN MEDICAL BIOLOGY. CELL STRUCTURE 1. Biology: The Science of Our Lives 2. Theories Contributing to Modern Biology: Cell Theory 3. Forms and Diversity of Life 4. Levels of Organization 5. Cell Structure Biology (from Greek βίος - life and λόγος - word, judgement) – is a branch of the natural sciences, and is the study of living organisms and their interactions with environment. The term was specially proposed by French naturalist Jean-Baptiste Pierre Antoine de Monet, Chevalier de Lamarck in 1802 Biology deals with every aspect of life in a living organism. Biology examines the structure, function, growth, origin, evolution, and distribution of living things. Modern biology is complex of sciences. Most biological sciences are specialized disciplines: Botany, Zoology, Protozoology, Microbiology, Virology, Molecular biology, Genetics, Embryology, Evolution theory, Ecology and so on. The history of biology traces the study of the living world from ancient to modern times. Although the concept of biology as a single coherent field arose in the 19th century, the biological sciences emerged from traditions of medicine and natural history reaching back to ancient Egyptian medicine and the works of Aristotle and Galen in the ancient Greco-Roman world, which were then further developed in the Middle Ages by Muslim physicians and scholars such as al-Jahiz, Avicenna, Avenzoar, Ibn al-Baitar and Ibn al-Nafis. Ancient Greek philosopher, Aristotle developed his Scala Naturae, or Ladder of Life, to explain his concept of the advancement -

Micrornas in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine
cells Review MicroRNAs in Prion Diseases—From Molecular Mechanisms to Insights in Translational Medicine Danyel Fernandes Contiliani 1,2, Yasmin de Araújo Ribeiro 1,2, Vitor Nolasco de Moraes 1,2 and Tiago Campos Pereira 1,2,* 1 Graduate Program of Genetics, Department of Genetics, Faculty of Medicine of Ribeirao Preto, University of Sao Paulo, Av. Bandeirantes, Ribeirao Preto 3900, Brazil; [email protected] (D.F.C.); [email protected] (Y.d.A.R.); [email protected] (V.N.d.M.) 2 Department of Biology, Faculty of Philosophy, Sciences and Letters, University of Sao Paulo, Av. Bandeirantes, Ribeirao Preto 3900, Brazil * Correspondence: [email protected]; Tel.: +55-16-3315-3818 Abstract: MicroRNAs (miRNAs) are small non-coding RNA molecules able to post-transcriptionally regulate gene expression via base-pairing with partially complementary sequences of target tran- scripts. Prion diseases comprise a singular group of neurodegenerative conditions caused by endoge- nous, misfolded pathogenic (prion) proteins, associated with molecular aggregates. In humans, classical prion diseases include Creutzfeldt–Jakob disease, fatal familial insomnia, Gerstmann– Sträussler–Scheinker syndrome, and kuru. The aim of this review is to present the connections between miRNAs and prions, exploring how the interaction of both molecular actors may help understand the susceptibility, onset, progression, and pathological findings typical of such disorders, as well as the interface with some prion-like disorders, such as Alzheimer’s. Additionally, due to the inter-regulation of prions and miRNAs in health and disease, potential biomarkers for non-invasive miRNA-based diagnostics, as well as possible miRNA-based therapies to restore the levels of dereg- Citation: Contiliani, D.F.; Ribeiro, Y.d.A.; de Moraes, V.N.; Pereira, T.C. -

Introduction to Viroids and Prions
Harriet Wilson, Lecture Notes Bio. Sci. 4 - Microbiology Sierra College Introduction to Viroids and Prions Viroids – Viroids are plant pathogens made up of short, circular, single-stranded RNA molecules (usually around 246-375 bases in length) that are not surrounded by a protein coat. They have internal base-pairs that cause the formation of folded, three-dimensional, rod-like shapes. Viroids apparently do not code for any polypeptides (proteins), but do cause a variety of disease symptoms in plants. The mechanism for viroid replication is not thoroughly understood, but is apparently dependent on plant enzymes. Some evidence suggests they are related to introns, and that they may also infect animals. Disease processes may involve RNA-interference or activities similar to those involving mi-RNA. Prions – Prions are proteinaceous infectious particles, associated with a number of disease conditions such as Scrapie in sheep, Bovine Spongiform Encephalopathy (BSE) or Mad Cow Disease in cattle, Chronic Wasting Disease (CWD) in wild ungulates such as muledeer and elk, and diseases in humans including Creutzfeld-Jacob disease (CJD), Gerstmann-Straussler-Scheinker syndrome (GSS), Alpers syndrome (in infants), Fatal Familial Insomnia (FFI) and Kuru. These diseases are characterized by loss of motor control, dementia, paralysis, wasting and eventually death. Prions can be transmitted through ingestion, tissue transplantation, and through the use of comtaminated surgical instruments, but can also be transmitted from one generation to the next genetically. This is because prion proteins are encoded by genes normally existing within the brain cells of various animals. Disease is caused by the conversion of normal cell proteins (glycoproteins) into prion proteins. -

The CPEB3 Ribozyme Modulates Hippocampal-Dependent Memory
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.23.426448; this version posted January 24, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The CPEB3 ribozyme modulates hippocampal-dependent memory Claire C. Chen1, Joseph Han2, Carlene A. Chinn2, Xiang Li2†, Mehran Nikan3, Marie Myszka4, Liqi Tong5, Timothy W. Bredy2†, Marcelo A. Wood2*, Andrej Lupták1,4,6* 1 Department of Pharmaceutical Sciences, University of California–Irvine, Irvine, California 92697, United States. 2 Department of Neurobiology and Behavior, Center for the Neurobiology of Learning and Memory, University of California–Irvine, Irvine, California 92697, United States. 3 Ionis Pharmaceuticals, 2855 Gazelle Court, Carlsbad, CA 92010, USA. 4 Department of Chemistry, University of California–Irvine, Irvine, California 92697, United States. 5 Institute for Memory Impairments and Neurological Disorders, University of California– Irvine, Irvine, California 92697, United States. 6 Department of Molecular Biology and Biochemistry, University of California–Irvine, Irvine, California 92697, United States *Correspondence to: Andrej Lupták. Department of Pharmaceutical Sciences, University of California–Irvine, Irvine, California 92697, United States. [email protected]. Marcelo A. Wood. Department of Neurobiology and Behavior, Center for the Neurobiology of Learning and Memory, University of California–Irvine, Irvine, California 92697, United States. [email protected]. † Present address: Cognitive Neuroepigenetics Laboratory, Queensland Brain Institute, The University of Queensland, Brisbane, QLD 4072, Australia. Keywords: ribozyme, splicing, self-scission, polyadenylation, local translation 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.23.426448; this version posted January 24, 2021. -

Mechanisms for Curing Yeast Prions
International Journal of Molecular Sciences Review Mechanisms for Curing Yeast Prions Lois E. Greene 1,*, Farrin Saba 1, Rebecca E. Silberman 1,2 and Xiaohong Zhao 1 1 Laboratory of Cell Biology, National Heart Lung Blood Institute National Institutes of Health, Bethesda, MD 20892, USA; [email protected] (F.S.); [email protected] (R.E.S.); [email protected] (X.Z.) 2 Koch Institute for Integrative Cancer Research, Depart Biol., Howard Hughes Medical Institute, MIT Cambridge, MA 02139, USA * Correspondence: [email protected]; Tel.: +1-301-496-1228 Received: 31 July 2020; Accepted: 2 September 2020; Published: 7 September 2020 Abstract: Prions are infectious proteins that self-propagate by changing from their normal folded conformation to a misfolded conformation. The misfolded conformation, which is typically rich in β-sheet, serves as a template to convert the prion protein into its misfolded conformation. In yeast, the misfolded prion proteins are assembled into amyloid fibers or seeds, which are constantly severed and transmitted to daughter cells. To cure prions in yeast, it is necessary to eliminate all the prion seeds. Multiple mechanisms of curing have been found including inhibiting severing of the prion seeds, gradual dissolution of the prion seeds, asymmetric segregation of the prion seeds between mother and daughter cells during cell division, and degradation of the prion seeds. These mechanisms, achieved by using different protein quality control machinery, are not mutually exclusive; depending on conditions, multiple mechanisms may work simultaneously to achieve curing. This review discusses the various methods that have been used to differentiate between these mechanisms of curing. -

Elucidating the Regulatory Mechanism of Swi1 Prion in Global Transcription
www.nature.com/scientificreports OPEN Elucidating the regulatory mechanism of Swi1 prion in global transcription and stress responses Zhiqiang Du1*, Jeniece Regan1, Elizabeth Bartom1, Wei‑Sheng Wu2, Li Zhang1,3, Dustin Kenneth Goncharof1 & Liming Li1* Transcriptional regulators are prevalent among identifed prions in Saccharomyces cerevisiae, however, it is unclear how prions afect genome‑wide transcription. We show here that the prion ([SWI+]) and mutant (swi1∆) forms of Swi1, a subunit of the SWI/SNF chromatin‑remodeling complex, confer dramatically distinct transcriptomic profles. In [SWI+] cells, genes encoding for 34 transcription factors (TFs) and 24 Swi1‑interacting proteins can undergo transcriptional modifcations. Several TFs show enhanced aggregation in [SWI+] cells. Further analyses suggest that such alterations are key factors in specifying the transcriptomic signatures of [SWI+] cells. Interestingly, swi1∆ and [SWI+] impose distinct and oftentimes opposite efects on cellular functions. Translation‑associated activities, in particular, are signifcantly reduced in swi1∆ cells. Although both swi1∆ and [SWI+] cells are similarly sensitive to thermal, osmotic and drought stresses, harmful, neutral or benefcial efects were observed for a panel of tested chemical stressors. Further analyses suggest that the environmental stress response (ESR) is mechanistically diferent between swi1∆ and [SWI+] cells—stress‑inducible ESR (iESR) are repressed by [SWI+] but unchanged by swi1∆ while stress‑repressible ESR (rESR) are induced by [SWI+] but repressed by swi1∆. Our work thus demonstrates primarily gain‑of‑function outcomes through transcriptomic modifcations by [SWI+] and highlights a prion‑mediated regulation of transcription and phenotypes in yeast. Te term “prions” was frst used to describe the proteinaceous pathogen of the fatal neurodegenerative diseases known as transmissible spongiform encephalopathies (TSEs) or prion diseases 1. -

RNA/Peptide Editing in Small Soluble Binding Proteins, a New Theory for the Origin of Life on Earth’S Crust
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 30 January 2020 RNA/peptide editing in small soluble binding proteins, a new theory for the origin of life on Earth’s crust Jean-François Picimbon QILU University of Technology, School of Bioengineering, Jinan 250353, Shandong, China Corresponding Author: Picimbon JF QILU University of Technology, School of Bioengineering Jinan 250353, Shandong, China Email: [email protected]/[email protected] Running Title: Evolutionary path from protocell to brain © 2020 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 30 January 2020 Abstract We remind about the dogma initially established with the nucleic acid double helix, i.e. the DNA structure as the primary source of life. However, we bring into the discussion those additional processes that were crucial to enable life and cell evolution. Studying chemosensory proteins (CSPs) and odor binding proteins (OBPs) of insects, we have found a high level of pinpoint mutations on the RNA and peptide sequences. Many of these mutations are found to be tissue-specific and induce subtle changes in the protein structure, leading to a new theory of cell multifunction and life evolution. Here, attention is given to RNA and peptide mutations in small soluble protein families known for carrying lipids and fatty acids as fuel for moth cells. A new phylogenetic analysis of mutations is presented and provides even more support to the pioneer work, i.e. the finding that mutations in binding proteins have spread through moths and various groups of insects. Then, focus is given to specific mechanisms of mutations that are not random, change -helical profilings and bring new functions at the protein level.