Dock3 Induces Axonal Outgrowth by Stimulating Membrane Recruitment of the WAVE Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
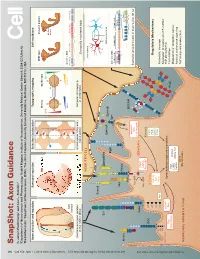
Snapshot: Axon Guidance Pasterkamp R
494 1 Cell Cell ??? SnapShot: Axon Guidance 153 SnapShot: XXXXXXXXXXXXXXXXXXXXXXXXXX 1 2 , ??MONTH?? ??DATE??, 200? ©200? Elsevier Inc. 200?©200? ElsevierInc. , ??MONTH?? ??DATE??, DOI R. Jeroen Pasterkamp and Alex L. Kolodkin , April11, 2013©2013Elsevier Inc. DOI http://dx.doi.org/10.1016/j.cell.2013.03.031 AUTHOR XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX 1 AFFILIATIONDepartment of XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, 3584 CG Utrecht, The Netherlands; 2Department of Neuroscience, HHMI, The Johns Hopkins University School of Medicine, Baltimore, MD 21212, USA Axon attraction and repulsion Surround repulsion Selective fasciculation Topographic mapping Self-avoidance Wild-type Dscam1 mutant Sema3A A Retina SC/Tectum EphB EphrinB P D D Mutant T A neuron N P A V V Genomic DNA P EphA EphrinA Slit Exon 4 (12) Exon 6 (48) Exon 9 (33) Exon 17 (2) Netrin Commissural axon guidance Surround repulsion of peripheral Grasshopper CNS axon Retinotectal mapping at the CNS midline nerves in vertebrates fasciculation in vertebrates Drosophila mushroom body XXXXXXXXX NEURITE/CELL Isoneuronal Sema3 Slit Sema1/4-6 Heteroneuronal EphrinA EphrinB FasII Eph Genomic DNA Pcdh-α (14) Pcdh-β (22) Pcdh-γ (22) Variable Con Variable Con Nrp Plexin ** *** Netrin LAMELLIPODIA Con DSCAM ephexin Starburst amacrine cells in mammalian retina Ras-GTP Vav See online version for legend and references. α-chimaerin GEFs/GAPs Robo FARP Ras-GDP LARG RhoGEF Kinases DCC cc0 GTPases PKA cc1 FAK Regulatory Mechanisms See online versionfor??????. Cdc42 GSK3 cc2 Rac PI3K P1 Rho P2 cc3 Abl Proteolytic cleavage P3 Regulation of expression (TF, miRNA, FILOPODIA srGAP Cytoskeleton regulatory proteins multiple isoforms) Sos Trio Pcdh Cis inhibition DOCK180 PAK ROCK Modulation of receptors’ output LIMK Myosin-II Colin Forward and reverse signaling Actin Trafcking and endocytosis NEURONAL GROWTH CONE Microtubules SnapShot: Axon Guidance R. -

Regulation of the Mammalian Target of Rapamycin Complex 2 (Mtorc2)
Regulation of the Mammalian Target Of Rapamycin Complex 2 (mTORC2) Inauguraldissertation Zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Klaus-Dieter Molle aus Heilbronn, Deutschland Basel, 2006 Genehmigt von der Philosophisch-Naturwissenschaftlichen Fakultät Auf Antrag von Prof. Michael N. Hall und Prof. Markus Affolter. Basel, den 21.11.2006 Prof. Hans-Peter Hauri Dekan Summary The growth controlling mammalian Target of Rapamycin (mTOR) is a conserved Ser/Thr kinase found in two structurally and functionally distinct complexes, mTORC1 and mTORC2. The tumor suppressor TSC1-TSC2 complex inhibits mTORC1 by acting on the small GTPase Rheb, but the role of TSC1-TSC2 and Rheb in the regulation of mTORC2 is unclear. Here we examined the role of TSC1-TSC2 in the regulation of mTORC2 in human embryonic kidney 293 cells. Induced knockdown of TSC1 and TSC2 (TSC1/2) stimulated mTORC2-dependent actin cytoskeleton organization and Paxillin phosphorylation. Furthermore, TSC1/2 siRNA increased mTORC2-dependent Ser473 phosphorylation of plasma membrane bound, myristoylated Akt/PKB. This suggests that loss of Akt/PKB Ser473 phosphorylation in TSC mutant cells, as reported previously, is due to inhibition of Akt/PKB localization rather than inhibition of mTORC2 activity. Amino acids and overexpression of Rheb failed to stimulate mTORC2 signaling. Thus, TSC1-TSC2 also inhibits mTORC2, but possibly independently of Rheb. Our results suggest that mTORC2 hyperactivation may contribute to the pathophysiology of diseases such as cancer and Tuberous Sclerosis Complex. i Acknowledgement During my PhD studies in the Biozentrum I received a lot of support from many people around me who I mention here to express my gratefulness. -

The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination
The Journal of Neuroscience, August 31, 2011 • 31(35):12579–12592 • 12579 Cellular/Molecular The Atypical Guanine-Nucleotide Exchange Factor, Dock7, Negatively Regulates Schwann Cell Differentiation and Myelination Junji Yamauchi,1,3,5 Yuki Miyamoto,1 Hajime Hamasaki,1,3 Atsushi Sanbe,1 Shinji Kusakawa,1 Akane Nakamura,2 Hideki Tsumura,2 Masahiro Maeda,4 Noriko Nemoto,6 Katsumasa Kawahara,5 Tomohiro Torii,1 and Akito Tanoue1 1Department of Pharmacology and 2Laboratory Animal Resource Facility, National Research Institute for Child Health and Development, Setagaya, Tokyo 157-8535, Japan, 3Department of Biological Sciences, Tokyo Institute of Technology, Midori, Yokohama 226-8501, Japan, 4IBL, Ltd., Fujioka, Gumma 375-0005, Japan, and 5Department of Physiology and 6Bioimaging Research Center, Kitasato University School of Medicine, Sagamihara, Kanagawa 252-0374, Japan In development of the peripheral nervous system, Schwann cells proliferate, migrate, and ultimately differentiate to form myelin sheath. In all of the myelination stages, Schwann cells continuously undergo morphological changes; however, little is known about their underlying molecular mechanisms. We previously cloned the dock7 gene encoding the atypical Rho family guanine-nucleotide exchange factor (GEF) and reported the positive role of Dock7, the target Rho GTPases Rac/Cdc42, and the downstream c-Jun N-terminal kinase in Schwann cell migration (Yamauchi et al., 2008). We investigated the role of Dock7 in Schwann cell differentiation and myelination. Knockdown of Dock7 by the specific small interfering (si)RNA in primary Schwann cells promotes dibutyryl cAMP-induced morpholog- ical differentiation, indicating the negative role of Dock7 in Schwann cell differentiation. It also results in a shorter duration of activation of Rac/Cdc42 and JNK, which is the negative regulator of myelination, and the earlier activation of Rho and Rho-kinase, which is the positive regulator of myelination. -

Related Malignant Phenotypes in the Nf1-Deficient MPNST
Published OnlineFirst February 19, 2013; DOI: 10.1158/1541-7786.MCR-12-0593 Molecular Cancer Genomics Research RAS/MEK–Independent Gene Expression Reveals BMP2- Related Malignant Phenotypes in the Nf1-Deficient MPNST Daochun Sun1, Ramsi Haddad2,3, Janice M. Kraniak2, Steven D. Horne1, and Michael A. Tainsky1,2 Abstract Malignant peripheral nerve sheath tumor (MPNST) is a type of soft tissue sarcoma that occurs in carriers of germline mutations in Nf1 gene as well as sporadically. Neurofibromin, encoded by the Nf1 gene, functions as a GTPase-activating protein (GAP) whose mutation leads to activation of wt-RAS and mitogen-activated protein kinase (MAPK) signaling in neurofibromatosis type I (NF1) patients' tumors. However, therapeutic targeting of RAS and MAPK have had limited success in this disease. In this study, we modulated NRAS, mitogen-activated protein/extracellular signal–regulated kinase (MEK)1/2, and neurofibromin levels in MPNST cells and determined gene expression changes to evaluate the regulation of signaling pathways in MPNST cells. Gene expression changes due to neurofibromin modulation but independent of NRAS and MEK1/2 regulation in MPNST cells indicated bone morphogenetic protein 2 (Bmp2) signaling as a key pathway. The BMP2-SMAD1/5/8 pathway was activated in NF1-associated MPNST cells and inhibition of BMP2 signaling by LDN-193189 or short hairpin RNA (shRNA) to BMP2 decreased the motility and invasion of NF1-associated MPNST cells. The pathway-specific gene changes provide a greater understanding of the complex role of neurofibromin in MPNST pathology and novel targets for drug discovery. Mol Cancer Res; 11(6); 616–27. -

A Rac/Cdc42 Exchange Factor Complex Promotes Formation of Lateral filopodia and Blood Vessel Lumen Morphogenesis
ARTICLE Received 1 Oct 2014 | Accepted 26 Apr 2015 | Published 1 Jul 2015 DOI: 10.1038/ncomms8286 OPEN A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis Sabu Abraham1,w,*, Margherita Scarcia2,w,*, Richard D. Bagshaw3,w,*, Kathryn McMahon2,w, Gary Grant2, Tracey Harvey2,w, Maggie Yeo1, Filomena O.G. Esteves2, Helene H. Thygesen2,w, Pamela F. Jones4, Valerie Speirs2, Andrew M. Hanby2, Peter J. Selby2, Mihaela Lorger2, T. Neil Dear4,w, Tony Pawson3,z, Christopher J. Marshall1 & Georgia Mavria2 During angiogenesis, Rho-GTPases influence endothelial cell migration and cell–cell adhesion; however it is not known whether they control formation of vessel lumens, which are essential for blood flow. Here, using an organotypic system that recapitulates distinct stages of VEGF-dependent angiogenesis, we show that lumen formation requires early cytoskeletal remodelling and lateral cell–cell contacts, mediated through the RAC1 guanine nucleotide exchange factor (GEF) DOCK4 (dedicator of cytokinesis 4). DOCK4 signalling is necessary for lateral filopodial protrusions and tubule remodelling prior to lumen formation, whereas proximal, tip filopodia persist in the absence of DOCK4. VEGF-dependent Rac activation via DOCK4 is necessary for CDC42 activation to signal filopodia formation and depends on the activation of RHOG through the RHOG GEF, SGEF. VEGF promotes interaction of DOCK4 with the CDC42 GEF DOCK9. These studies identify a novel Rho-family GTPase activation cascade for the formation of endothelial cell filopodial protrusions necessary for tubule remodelling, thereby influencing subsequent stages of lumen morphogenesis. 1 Institute of Cancer Research, Division of Cancer Biology, 237 Fulham Road, London SW3 6JB, UK. -

Anti-CRK Monoclonal Antibody, Clone 4H22D2 (DCABH-1226) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-CRK monoclonal antibody, clone 4H22D2 (DCABH-1226) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Mouse monoclonal to Crk p38 Antigen Description The Crk-I and Crk-II forms differ in their biological activities. Crk-II has less transforming activity than Crk-I. Crk-II mediates attachment-induced MAPK8 activation, membrane ruffling and cell motility in a Rac-dependent manner. Involved in phagocytosis of apoptotic cells and cell motility via its interaction with DOCK1 and DOCK4. Immunogen Purified recombinant fragment of Human Crk p38 expressed in E. Coli. Isotype IgG2b Source/Host Mouse Species Reactivity Mouse, Human Clone 4H22D2 Purity Ascites Conjugate Unconjugated Applications WB, ELISA, IHC-P, Flow Cyt, ICC/IF Positive Control Recombinant Crk p38 protein; Crk p38-hIgGFc transfected HEK293 cell lysate; Human rectum cancer and bladder cancer tissues; MCF7 and 3T3 L1 cells. Format Liquid Size 100 μl Buffer Preservative: 0.03% Sodium azide; Constituent: 99% Ascites Preservative 0.03% Sodium Azide Storage Store at 4°C or at -20°C for long term storage. GENE INFORMATION 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Gene Name CRK v-crk sarcoma virus CT10 oncogene homolog (avian) [ Homo sapiens ] Official Symbol CRK Synonyms CRK; v-crk sarcoma virus CT10 oncogene homolog (avian); v crk avian sarcoma virus CT10 oncogene homolog; adapter molecule crk; proto-oncogene -

Targeting PH Domain Proteins for Cancer Therapy
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2018 Targeting PH domain proteins for cancer therapy Zhi Tan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Medicinal Chemistry and Pharmaceutics Commons, Neoplasms Commons, and the Pharmacology Commons Recommended Citation Tan, Zhi, "Targeting PH domain proteins for cancer therapy" (2018). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 910. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/910 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. TARGETING PH DOMAIN PROTEINS FOR CANCER THERAPY by Zhi Tan Approval page APPROVED: _____________________________________________ Advisory Professor, Shuxing Zhang, Ph.D. _____________________________________________ -

Crit. Rev. in Biochem. and Molec. Biol. 2011
Critical Reviews in Biochemistry and Molecular Biology Critical Reviews in Biochemistry and Molecular Biology, 2011, 1–21, Early Online 2011 © 2011 Informa Healthcare USA, Inc. ISSN 1040-9238 print/ISSN 1549-7798 online 00 DOI: 10.3109/10409238.2011.618113 00 000 REVIEW 000 18 June 2011 Mammalian TOR signaling to the AGC kinases 15 August 2011 Bing Su1 and Estela Jacinto2 24 August 2011 1Department of Immunobiology and The Vascular Biology and Therapeutics Program, Yale University School of Medicine, New Haven, CT, USA and 2Department of Physiology and Biophysics, UMDNJ-RWJMS, Piscataway, NJ, USA 1040-9238 1549-7798 Abstract The mechanistic (or mammalian) target of rapamycin (mTOR), an evolutionarily conserved protein kinase, orchestrates © 2011 Informa Healthcare USA, Inc. cellular responses to growth, metabolic and stress signals. mTOR processes various extracellular and intracellular inputs as part of two mTOR protein complexes, mTORC1 or mTORC2. The mTORCs have numerous cellular targets but 10.3109/10409238.2011.618113 members of a family of protein kinases, the protein kinase (PK)A/PKG/PKC (AGC) family are the best characterized direct mTOR substrates. The AGC kinases control multiple cellular functions and deregulation of many members of BBMG this family underlies numerous pathological conditions. mTOR phosphorylates conserved motifs in these kinases to allosterically augment their activity, influence substrate specificity, and promote protein maturation and 618113 stability. Activation of AGC kinases in turn triggers the phosphorylation of diverse, often overlapping, targets that ultimately control cellular response to a wide spectrum of stimuli. This review will highlight recent findings on how mTOR regulates AGC kinases and how mTOR activity is feedback regulated by these kinases. -

A Rhog-Mediated Signaling Pathway That Modulates Invadopodia Dynamics in Breast Cancer Cells Silvia M
© 2017. Published by The Company of Biologists Ltd | Journal of Cell Science (2017) 130, 1064-1077 doi:10.1242/jcs.195552 RESEARCH ARTICLE A RhoG-mediated signaling pathway that modulates invadopodia dynamics in breast cancer cells Silvia M. Goicoechea, Ashtyn Zinn, Sahezeel S. Awadia, Kyle Snyder and Rafael Garcia-Mata* ABSTRACT micropinocytosis, bacterial uptake, phagocytosis and leukocyte One of the hallmarks of cancer is the ability of tumor cells to invade trans-endothelial migration (deBakker et al., 2004; Ellerbroek et al., surrounding tissues and metastasize. During metastasis, cancer cells 2004; Jackson et al., 2015; Katoh et al., 2006, 2000; van Buul et al., degrade the extracellular matrix, which acts as a physical barrier, by 2007). Recent studies have revealed that RhoG plays a role in tumor developing specialized actin-rich membrane protrusion structures cell invasion and may contribute to the formation of invadopodia called invadopodia. The formation of invadopodia is regulated by Rho (Hiramoto-Yamaki et al., 2010; Kwiatkowska et al., 2012). GTPases, a family of proteins that regulates the actin cytoskeleton. Invadopodia are actin-rich adhesive structures that form in the Here, we describe a novel role for RhoG in the regulation of ventral surface of cancer cells and allow them to degrade the invadopodia disassembly in human breast cancer cells. Our results extracellular matrix (ECM) (Gimona et al., 2008). Formation of show that RhoG and Rac1 have independent and opposite roles invadopodia involves a series of steps that include the disassembly in the regulation of invadopodia dynamics. We also show that SGEF of focal adhesions and stress fibers, and the relocalization of several (also known as ARHGEF26) is the exchange factor responsible of their components into the newly formed invadopodia (Hoshino for the activation of RhoG during invadopodia disassembly. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

Identification of Two Signaling Submodules Within the Crkii/ELMO
Cell Death and Differentiation (2007) 14, 963–972 & 2007 Nature Publishing Group All rights reserved 1350-9047/07 $30.00 www.nature.com/cdd Identification of two signaling submodules within the CrkII/ELMO/Dock180 pathway regulating engulfment of apoptotic cells A-C Tosello-Trampont1,4, JM Kinchen1,2,4, E Brugnera1,3, LB Haney1, MO Hengartner2 and KS Ravichandran*,1 Removal of apoptotic cells is a dynamic process coordinated by ligands on apoptotic cells, and receptors and other signaling proteins on the phagocyte. One of the fundamental challenges is to understand how different phagocyte proteins form specific and functional complexes to orchestrate the recognition/removal of apoptotic cells. One evolutionarily conserved pathway involves the proteins cell death abnormal (CED)-2/chicken tumor virus no. 10 (CT10) regulator of kinase (Crk)II, CED-5/180 kDa protein downstream of chicken tumor virus no. 10 (Crk) (Dock180), CED-12/engulfment and migration (ELMO) and MIG-2/RhoG, leading to activation of the small GTPase CED-10/Rac and cytoskeletal remodeling to promote corpse uptake. Although the role of ELMO : Dock180 in regulating Rac activation has been well defined, the function of CED-2/CrkII in this complex is less well understood. Here, using functional studies in cell lines, we observe that a direct interaction between CrkII and Dock180 is not required for efficient removal of apoptotic cells. Similarly, mutants of CED-5 lacking the CED-2 interaction motifs could rescue engulfment and migration defects in CED-5 deficient worms. Mutants of CrkII and Dock180 that could not biochemically interact could colocalize in membrane ruffles.