Experience with a Uranyl Nitrate/Uranium Dioxide Conversion Pilot Plant
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction Processing of Concentrated Solutions of Uranyl Nitrate with High Impurities Content V.M
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Electronic archive of Tomsk Polytechnic University Сhemistry UDC 546.791.02.238:66.061.51 EXTRACTION PROCESSING OF CONCENTRATED SOLUTIONS OF URANYL NITRATE WITH HIGH IMPURITIES CONTENT V.M. Korotkevich, V.V.Lazarchuck, T.G. Shikerun, V.I. Shamin, N.A. Mikhailova, F.A. Dorda Federal Unitary Enterprise Siberian Group of Chemical Enterprises, Seversk Еmail: [email protected] Process flowsheet of recycling uranium concentrated solutions with its purification from insoluble impurities of iron, silicon, molybde num, calcium oxides and hydroxides and soluble impurities with application of centrifugal extractors cascade has been developed and suggested for commercial introduction. The process was carried out at extractant saturation (30 % tributyl phosphate in hydrocarbon diluent) in extraction assembly lower than a limiting level (85...95 g/l) and in wash assembly – at limiting saturation (up to 120 g/l). As a result the waste uranium content in watertail solutions 0,01...0,04 g/l and minimal content of impurities in reextractors is provided. Perspective program on development of nuclear technology are also: reliability of hermetization, possi power engineering proposes to involve wide range of raw bility of remote service, resistance to corrosive attack of materials from uranium ores to secondary uranium ma structural materials, duration of overhaul period etc. terials into processing at atomic enterprises. In this case In Russia the centrifugal extractor of ECТseries the problem of reprocessing concentrated uranium so with continuous extraction of solid phase [3], which may lutions with high impurity content occurs. -
Nitrate (UO2 (NO)) 4
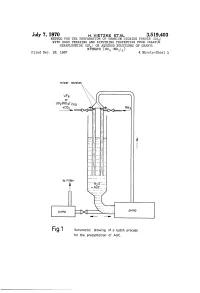
July 7, 1970 H. VETZKE ETA 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Fied Dec. 18, 1967 NiTRATE (UO2 (NO)) 4. Sheets-Sheet l mixer nozzles --race to fitter Fig.1 Schematic drawing of a batch process for the precipitation of AUC. July 7, 1970 H. VETZKE EA 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM RANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Filed Dec. 8, 1967 NiTRATE (UO2 (NO)) 4. Sheets-Sheet 3. mixer nozzles UF6 Of U02 (NO3)2ad tC92--X Ex-Ns / to filter cooling precipitation Vessel vessel Fig. 2 Schematic drawing of a continuous process for the precipitation of AUC July 7, 1970 H, VETZKE ETAL 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Filed Dec. 18, 1967 NiTRATE (UO (NO)2) 4. Sheets-Sheet 3 Off gas Off gas 487\m Nx UO2 powder XXX.& X.X S& steam / H2 Fig. 3 Ot Schematic drawing of a fluidized bed furnace for the reduction of AUC to U02, July 7, 1970 H. VEZKE ETAL 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL NITRATE (UO2 (NO)a) Filed Dec. 18, 1967 4. Sheets-Sheet 1 -- Offgas Step 1: Decomposition, reduction and pyrohydrolysis i Step 2: Pyrohydrolysis Step 3: Controlled oxidation an o- are as an un- Fig. -

United States Pmao" ICC Patented June 30, '1959 1 2 Adsorption on Manganese Dioxide, but It May on the 2,892,677 Other Hand He Left in the Solution
r. 2,892,677 United States Pmao" ICC Patented June 30, '1959 1 2 adsorption on manganese dioxide, but it may on the 2,892,677 other hand he left in the solution. If it is removed, a certain amount of manganese nitrate is formed from the ' SEPARATION OF URANIUM FROM THORIUM manganese dioxide in the solution and this nitrate reacts AND PROTACTINIUM with the sodium diethyldithiocarbamate to give manga William Kenneth Rodgerson Musgrave, Durham, Eng ' nese diethyldithiocarbamate, which must subsequently be land, assignor, by mesne assignments, to the United separated from the uranium. ,, . .States of America as represented by the United States Whether or not the protactinium has been removed, Atomic Energy Commission the solution is brought to a pH of between 2 and 3, for No Drawing. Application November 27, 1946 10 example by the addition of ammonia. The sodium di Serial No. 712,722 ethyldithiocarbamate is then dissolved in a solvent in which the subsequently formed uranyl diethyldithiocar '8 Claims. (Cl. 23—14.5) bamate is also soluble. Such a solvent is, for example, amyl acetate or methyl isobutyl ketone, the former being This invention relates to the separation of uranium 15 preferable for the reasons which will be indicated below. from thorium and protactinium. A mixture of these The sodium diethyldithiocarbamate is dissolved in amyl elements is obtained, for example, as the result of irradia acetate to form a solution containing 0.25% of the tion by neutrons of so-called thorium carbonate, which former, and this solution is then shaken up with the is a mixture of thorium oxide and thorium carbonate. -

Solvent Extraction of Uranium with Acetylacetone and Tri-N-Butyl
DAEHAN HWAHAK HWOEJEE (Journal of the Korean Chemical Society) Vol. 24, No. 3, 1980 Printed in the Republic of Korea 아세 틸아세톤과 트리부틸인산의 도데칸용액 에 의한 우라늄의 용매추출 裴奎善-鄰奇碩 韓國核燃料開發公團 (1980. 2. 27 接受 ) Solvent Extraction of Uranium with Acetylacetone and Tri-n-Butyl Phosphate in “-Dodecane Kyu Sun Bai and Key Suck Jung Korea Nuclear Fuel Development Institute, Daeduck 300-32, Korea (Received Feb. 27, 1980) 요 악 . 도데칸에 녹인 아세틸아세톤과 트리부틸인산으로 묽은 질산우라닐수용액에서 우라늄 (VI) 을 추출했다 . pHl 이상에서 이 혼합추출제의 상승적 효과가 관측되었다 . 추출되는 화학종은 1:2:1 및 1:2:2 우라닐 -아세털 아세톤 -트리부털 인산착물이다 . 이들 반응의 추출정수들을 측정하였다 . ABSTRACT. Uranium (VI) was extracted from dilute aqueous solutions of uranyl nitrate with acetylacetone and tri-n-butyl phosphate in n-dodecane. Synergistic effect was observed with the mixed reagents above pH 1, The species extracted are the 1:2:1 and the 1:2:2 uranyl-AA-TBP complexes. The extraction constants for these reactions have been determined. by crystallization of the salt as UO2(NO3)2 INTRODUCTION •6H2O. HAA and TBP were dissolved in do Acetylacetone (HAA) is a simple -diketone. decane and uranyl nitrate in water. Equal A weak acid with pKa=8. 2% HAA has long volumes of organic and aqueous phases were been used as a chelating agent for many equilibrated in 100 mZ separatory funnels by metals.2 It is quite soluble in water and its vigorous shaking for several minutes. Equili brium temperature was 25 °C or ambient. pH distribution coefficient is E=5. 95 for benzene/ water system.3 Because of its solubility in measurements were made by using a Corning water, HAA has not been so widely used as, Model 130 pH meter. -

Material Safety Data Sheet
SAFETY DATA SHEET URANYL NITRATE SOLUTION SECTION 1: CHEMICAL PRODUCT & COMPANY IDENTIFICATION New Brunswick Laboratory U.S. Department of Energy 9800 South Cass Avenue Argonne, IL 60439 1-630-252-CRMS (2767) Emergency Phone Numbers: 1-630-252-6130 or 1-630-252-5731 Chemical Name: Uranyl Nitrate Solution, UO2(NO3)2 Other Identifiers: Certified Reference Material (CRM) standard or Safeguards Measurement Evaluation (SME) sample Use and Restrictions: This material is prepared for use as a standard or in inter-laboratory comparison programs at analytical laboratories, which routinely handle uranium and/or plutonium. NBL expects that recipients of their material are in compliance with 29 CFR 1910.1200(h) which requires employers to provide employees with effective information and training on hazardous chemicals in their work area. SECTION 2: HAZARDS IDENTIFICATION OSHA HAZARDS: Skin Corrosion/Irritation Category 1B. Serious Eye Damage/Irritation Category 1 TARGET ORGANS: Skin, Eyes, Kidneys. GHS Label elements, including precautionary statements Signal Word: DANGER Pictogram: Page 1 of 12 Hazard Statement(s) H314 Causes severe skin burns and eye damage H373 May cause damage to organs through prolonged or repeated exposure. H411 Toxic to aquatic life with long lasting effects. Precautionary statement(s) P260 Do not breathe dust/ fume/ gas/ mist/ vapors/ spray. P262 Do not get in eyes, on skin, or on clothing. P264 Wash skin thoroughly after handling. P273 Avoid release to the environment. P280 Wear protective gloves/protective clothing/eye protection/face protection P310 Immediately call a POISON CENTER or doctor/ physician if swallowed or inhaled. Other Hazard(s): Radioactive NFPA RATINGS (SCALE 0-4): Health=3 Fire=0 Reactivity=0 Special Hazard= OX SECTION 3: COMPOSITION/INFORMATION ON INGREDIENTS Chemical Name: Uranyl Nitrate Solution Common Names/Synonyms: CRM U045; CRM 135; CRM 145; CRM 145-B; CRM U930-D; Safeguards Measurement Evaluation (SME) Low Enrichment, Normal Enrichment, or High Enrichment Solutions; Uranyl Nitrate in Nitric Acid Solution. -

Uranyl Nitrate, 1%
Uranyl Nitrate, 1% 1. Identification Product Name: Uranyl Nitrate, 1% Item #: DI0103014, DI0103029, SKC1048-250, SKC1048-500 Web SDS: S231 Synonyms: N/A Recommended Use: Special Stains Restrictions on Use: N/A Manufacturer: In Case of Emergency: BBC Biochemical Chemtrec US 1-800-424-9300 409 Eleanor Lane, Chemtrec International 703-527-3887 Mount Vernon, WA 98273 1-800-635-4477 2. Hazards Identification OSHA Hazard Classification(s): Acute Toxicity - Oral - Category 4 Acute Toxicity - Inhalation - Category 4 Signal Word: Warning Hazard Statement(s): Harmful if swallowed. Harmful if inhaled. Pictogram(s): Precautionary Statement(s): Prevention: Wash body thoroughly after handling. Do not eat, drink or smoke when using this product. Avoid breathing dust, vapors. Use only outdoors or in a well-ventilated area. Response: If swallowed: Call a doctor if you feel unwell. Rinse mouth. If inhaled: Remove person to fresh air and keep comfortable for breathing. Call a doctor if you feel unwell. Storage: N/A Disposal: Dispose of contents/container in accordance with local regulations. Descriptions of Hazards not otherwise classified: N/A Percent of mixture with unknown acute toxicity: N/A 3. Composition and Information on Ingredients Chemical Name Common Name CAS # Concentration % Uranyl Nitrate 13520-83-7 1 Water 7732-18-5 99 4. First Aid Measures Eye Contact: If in eyes: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. If eye irritation persists: Get medical advice/attention. Skin Contact: If on skin (or hair): Take off immediately all contaminated clothing and wash before reuse. -

Physcio-Chemical Studies on the Systems Uranyl Nitrate Organic Solvent
Durham E-Theses Physcio-chemical studies on the systems uranyl nitrate organic solvent - water Mathieson A. R., How to cite: Mathieson A. R., (1951) Physcio-chemical studies on the systems uranyl nitrate organic solvent - water, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/9766/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk PSYSIC0-CH3MICAL STUDIES OH THE SYSTEMS DEAUTL 1TITRATE - ORGANIC SOLVENT - WATER A.R. Mathieson, B.Sc.(Dunelm), A.R.I.C. Thesis presented for the degree of M. Sc. in Pure Science of the University of Durham, August 1951 The experimental work described in this thesis was performed at the Atomic Energy Research Establishment, Harwell, during the period November 1947 - December 1949* The results have already been published elsewhere: Mathieson "Stability of Complexes of Uranyl Nitrate with Ketones and Ethers", J. -

Analysis of Mo Production Capacity in Uranyl Nitrate Aqueous
A. Isnaeni,Atom atIndonesia al., / Atom Vol. Indonesia 40 No. 1 Vol.(2014) 40 40No. - 143 (2014) Analysis of 99Mo Production Capacity in Uranyl Nitrate Aqueous Homogeneous Reactor using ORIGEN and MCNP A. Isnaeni*, M.S. Aljohani, T.G. Aboalfaraj and S.I. Bhuiyan Nuclear Engineering Department, King Abdulaziz University P.O. Box 80240 Jeddah, Kingdom of Saudi Arabia A R T I C L E I N F O A B S T R A C T Article history: 99mTc is a very useful radioisotope in medical diagnostic procedure. 99mTc is Received 11 January 2014 produced from 99Mo decay. Currently, most of 99Mo is produced by irradiating 235U Received in revised form 18 February 2014 in the nuclear reactor. 99Mo mostly results from the fission reaction of 235U targets Accepted 28 February 2014 with a fission yield about 6.1%. A small additional amount is created from 98Mo 99 neutron activation. Actually Mo is also created in the reactor fuel, but usually we Keywords: 99 do not extract it. The fuel will become spent fuel which is a highly radioactive Mo waste. 99Mo production system in the aqueous homogeneous reactor offers a better Uranyl nitrate method, because all of the 99Mo can be extracted from the fuel solution. Fresh Homogeneous reactor reactor fuel solution consists of uranyl nitrate dissolved in water. There is no MCNP separation of target and fuel in an aqueous homogeneous reactor where target and ORIGEN fuel become one liquid solution, and there is no spent fuel generated from this reactor. Simulation of the extraction process is performed while reactor in operation (without reactor shutdown). -

Uranyl Nitrate Hazard Summary Identification
Common Name: URANYL NITRATE CAS Number: 10102-06-4 RTK Substance number: 1978 DOT Number: UN 2981 (Solid) Date: April 2000 ----------------------------------------------------------------------- ----------------------------------------------------------------------- HAZARD SUMMARY * Uranyl Nitrate can affect you when breathed in and may * If you think you are experiencing any work-related health be absorbed through the skin. problems, see a doctor trained to recognize occupational * Contact can irritate and burn the skin and eyes with diseases. Take this Fact Sheet with you. possible eye damage. * Breathing Uranyl Nitrate can irritate the nose, throat and WORKPLACE EXPOSURE LIMITS lungs causing coughing, wheezing and/or shortness of The following exposure limits are for soluble Uranium breath. compounds (measured as Uranium): * High exposures to Uranyl Nitrate can cause nausea, vomiting, muscle weakness, and convulsions. OSHA: The legal airborne permissible exposure limit * Uranyl Nitrate may damage the liver and kidneys. (PEL) is 0.05 mg/m3 averaged over an 8-hour * Repeated exposure to Uranyl Nitrate may affect the brain. workshift. * CONSULT THE NEW JERSEY DEPARTMENT OF HEALTH AND SENIOR SERVICES HAZARDOUS NIOSH: The recommended airborne exposure limit is SUBSTANCE FACT SHEET ON URANIUM. 0.05 mg/m3 averaged over a 10-hour workshift. IDENTIFICATION ACGIH: The recommended airborne exposure limit is Uranyl Nitrate is a pale yellow, radioactive solid with no 0.2 mg/m3 averaged over an 8-hour workshift odor. It is used in photographic processes, glazing ceramics, and 0.6 mg/m3 as a STEL (short term exposure and radiation shielding. limit). REASON FOR CITATION * The above exposure limits are for air levels only. When * Uranyl Nitrate is on the Hazardous Substance List skin contact also occurs, you may be overexposed, even because it is regulated by OSHA and cited by ACGIH, though air levels are less than the limits listed above. -

Ammonium Diuranate' Prepared from Uranium Hexa¯Uoride J.A
L Journal of Alloys and Compounds 323±324 (2001) 838±841 www.elsevier.com/locate/jallcom Recovery of uranium from the ®ltrate of `ammonium diuranate' prepared from uranium hexa¯uoride J.A. Seneda* , F.F. Figueiredo, A. Abrao,Ä F.M.S. Carvalho, E.U.C. Frajndlich Environmental and Chemical Center, Instituto de Pesquisas Energeticas e Nucleares, Trav.R, 400, SaoÄ Paulo, CEP 05508-900, Brazil Abstract The hydrolysis of uranium hexa¯uoride and its conversion to `ammonium diuranate' yields an alkaline solution containing ammonium ¯uoride and low concentrations of uranium. The recovery of the uranium has the advantage of saving this valuable metal and the avoidance of unacceptable discarding the above mentioned solution to the environment. The recovery of uranium(VI) is based on its complex with the excess of ¯uoride in the solution and its adsorption on to an anionic ion-exchange resin. The `ammonium diuranate' ®ltrate has an approximate concentration of 130 mg l21 of uranium and 20 g l21 of ammonium ¯uoride. An effective separation and recovery of the uranyl ¯uoride was achieved by choosing a suitable pH and ¯ow rate of the uranium-bearing solution on to the resin column. The ef¯uent ammonium ¯uoride will be recovered as well. Uranium ¯uoride adsorbed by the ion-exchanger is then transformed into the corresponding uranyl tricarbonate complex by percolation of a dilute ammonium carbonate solution. Finally, the free ¯uoride uranium carbonate is eluted from the resin with a more concentrate ammonium carbonate solution. The eluate now can be storage to be precipitated as ammonium uranyl carbonate (AUC). -

1% Uranyl Nitrate
Page 1/8 SAFETY DATA SHEET In Accordance with ISO/DIS 11014 Review Date 04/06/2015 Revision Date 08/09/2012 1 Identification of the Substance/Mixture and Company Product Name: 1% Uranyl Nitrate MSDS Code: SSAHUNI1 Product Description: Laboratory Reagent Manufacturer/Supplier: American MasterTech 1330 Thurman Street Lodi, CA 95240 USA (800) 860-4073 European Authorized Representative: Emergo Europe Moleensraat 15 2513 BH The Hague, The Netherlands Emergency Telephone Number: Infotrac (800) 535-5053 (24 hours) - International (011) 352-323-3500 2 Hazards Identification Classification of the substance or mixture GHS07 Acute Tox. 4 H302 Harmful if swallowed. Acute Tox. 5 H333 May be harmful if inhaled. Classification according to Directive 67/548/EEC or Directive 1999/45/EC T; Toxic R23/25: Toxic by inhalation and if swallowed. R33: Danger of cumulative effects. Information concerning particular hazards for human and environment: The product has to be labelled due to the calculation procedure of international guidelines. Classification system: The classification was made according to the latest editions of international substances lists, and expanded upon from company and literature data. Label elements GHS label elements The product is classified and labeled according to the Globally Harmonized System (GHS). Hazard pictograms GHS07 Signal word Warning USA 34.2.6 Page 2/8 SAFETY DATA SHEET In Accordance with ISO/DIS 11014 Review Date 04/06/2015 Revision Date 08/09/2012 Product Name: 1% Uranyl Nitrate Hazard-determining components of labeling: Uranylnitrat-Hexahydrat Hazard statements Harmful if swallowed. May be harmful if inhaled. Precautionary statements If medical advice is needed, have product container or label at hand. -

Design of a Small Aqueous Homogeneous Reactor for Production of 99Mo Improving the Reliability of the Supply Chain
Design of a small Aqueous Homogeneous Reactor for production of 99Mo Improving the reliability of the supply chain S. Rijnsdorp Master Thesis DESIGNOFASMALL AQUEOUS HOMOGENEOUS REACTOR FOR PRODUCTION OF 99MO IMPROVING THE RELIABILITY OF THE SUPPLY CHAIN by S. Rijnsdorp BSc in partial fulfillment of the requirements for the degree of Master of Science in Applied Physics at Delft University of Technology, to be defended on Tuesday September 2, 2014 at 09:30. faculty Applied Sciences department Radiation Science and Technology section Nuclear Energy and Radiation Applications supervisors Dr.ir. J.L. Kloosterman Dr.ir. M. Rohde thesis committee Dr.ir. J.L. Kloosterman TU Delft, Applied Sciences, RST, NERA Dr.ir. M. Rohde TU Delft, Applied Sciences, RST, NERA Dr. Eng. L.M. Portela TU Delft, Applied Sciences, ChemE, TP ABSTRACT Medical isotopes are used worldwide for medical imaging and treatment of patients with different kinds of diseases, for example thyroid diseases and blood disorders. The most widely used radio- isotope for medical imaging is 99mTc, a daughter nuclide of 99Mo. Annually, around 400,000 diag- nostical treatments using 99mTc are carried out in the Netherlands. The characteristic aspect that makes 99mTc so useful for medical imaging is also the challenge: 99mTc has a half-life of approximately 6 hours. Because also the parent nuclide 99Mo has a relatively short half-life (66 hours), a continuous production and supply is needed. Recently, a number of events occurred, disrupting the supply, which led to the wish for a more reliable production method of radioisotopes. Currently, the entire world demand is produced in 5 reactors, meaning that an unplanned outage of one or more reactors will immediately decrease the supply of 99Mo.