Laboratory and Pilot-Plant Studies on the Conversion of Uranyl Nitrate Hexahydrate to Ufsby Fluidized-Bed Processes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction Processing of Concentrated Solutions of Uranyl Nitrate with High Impurities Content V.M
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Electronic archive of Tomsk Polytechnic University Сhemistry UDC 546.791.02.238:66.061.51 EXTRACTION PROCESSING OF CONCENTRATED SOLUTIONS OF URANYL NITRATE WITH HIGH IMPURITIES CONTENT V.M. Korotkevich, V.V.Lazarchuck, T.G. Shikerun, V.I. Shamin, N.A. Mikhailova, F.A. Dorda Federal Unitary Enterprise Siberian Group of Chemical Enterprises, Seversk Еmail: [email protected] Process flowsheet of recycling uranium concentrated solutions with its purification from insoluble impurities of iron, silicon, molybde num, calcium oxides and hydroxides and soluble impurities with application of centrifugal extractors cascade has been developed and suggested for commercial introduction. The process was carried out at extractant saturation (30 % tributyl phosphate in hydrocarbon diluent) in extraction assembly lower than a limiting level (85...95 g/l) and in wash assembly – at limiting saturation (up to 120 g/l). As a result the waste uranium content in watertail solutions 0,01...0,04 g/l and minimal content of impurities in reextractors is provided. Perspective program on development of nuclear technology are also: reliability of hermetization, possi power engineering proposes to involve wide range of raw bility of remote service, resistance to corrosive attack of materials from uranium ores to secondary uranium ma structural materials, duration of overhaul period etc. terials into processing at atomic enterprises. In this case In Russia the centrifugal extractor of ECТseries the problem of reprocessing concentrated uranium so with continuous extraction of solid phase [3], which may lutions with high impurity content occurs. -

Separation of Fluoride Residue Arising from Fluoride Volatility Recovery of Uranium from Spent Nuclear Fuel
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 5-2004 Separation of Fluoride Residue Arising from Fluoride Volatility Recovery of Uranium from Spent Nuclear Fuel Jennifer L. Ladd-Lively University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Chemical Engineering Commons Recommended Citation Ladd-Lively, Jennifer L., "Separation of Fluoride Residue Arising from Fluoride Volatility Recovery of Uranium from Spent Nuclear Fuel. " Master's Thesis, University of Tennessee, 2004. https://trace.tennessee.edu/utk_gradthes/2557 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Jennifer L. Ladd-Lively entitled "Separation of Fluoride Residue Arising from Fluoride Volatility Recovery of Uranium from Spent Nuclear Fuel." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Chemical Engineering. Robert M. Counce, Major Professor We have read this thesis and recommend its acceptance: Barry B. Spencer, Paul Bienkowski, Fred Weber Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) To the Graduate Council: I am submitting herewith a thesis written by Jennifer L. -
Nitrate (UO2 (NO)) 4
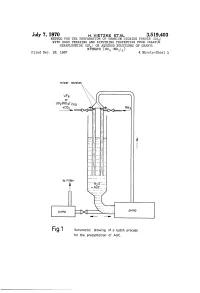
July 7, 1970 H. VETZKE ETA 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Fied Dec. 18, 1967 NiTRATE (UO2 (NO)) 4. Sheets-Sheet l mixer nozzles --race to fitter Fig.1 Schematic drawing of a batch process for the precipitation of AUC. July 7, 1970 H. VETZKE EA 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM RANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Filed Dec. 8, 1967 NiTRATE (UO2 (NO)) 4. Sheets-Sheet 3. mixer nozzles UF6 Of U02 (NO3)2ad tC92--X Ex-Ns / to filter cooling precipitation Vessel vessel Fig. 2 Schematic drawing of a continuous process for the precipitation of AUC July 7, 1970 H, VETZKE ETAL 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Filed Dec. 18, 1967 NiTRATE (UO (NO)2) 4. Sheets-Sheet 3 Off gas Off gas 487\m Nx UO2 powder XXX.& X.X S& steam / H2 Fig. 3 Ot Schematic drawing of a fluidized bed furnace for the reduction of AUC to U02, July 7, 1970 H. VEZKE ETAL 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL NITRATE (UO2 (NO)a) Filed Dec. 18, 1967 4. Sheets-Sheet 1 -- Offgas Step 1: Decomposition, reduction and pyrohydrolysis i Step 2: Pyrohydrolysis Step 3: Controlled oxidation an o- are as an un- Fig. -

Properties of Selected Radioisotopes
CASE FILE COPY NASA SP-7031 Properties of Selected Radioisotopes A Bibliography PART I: UNCLASSIFIED LITERATURE NATIONAL AERONAUTICS AND SPACE ADMINISTRATION NASA SP-7031 PROPERTIES OF SELECTED RADIOISOTOPES A Bibliography Part I: Unclassified Literature A selection of annotated references to technical papers, journal articles, and books This bibliography was compiled and edited by DALE HARRIS and JOSEPH EPSTEIN Goddard Space Flight Center Greenbelt, Maryland Scientific and Technical Information Division / OFFICE OF TECHNOLOGY UTILIZATION 1968 USP. NATIONAL AERONAUTICS AND SPACE ADMINISTRATION Washington, D.C. PREFACE The increasing interest in the application of substantial quantities of radioisotopes for propulsion, energy conversion, and various other thermal concepts emphasizes a need for the most recent and most accurate information available describing the nuclear, chemical, and physical properties of these isotopes. A substantial amount of progress has been achieved in recent years in refining old and developing new techniques of measurement of the properties quoted, and isotope processing. This has resulted in a broad technological base from which both the material and information about the material is available. Un- fortunately, it has also resulted in a multiplicity of sources so that information and data are either untimely or present properties without adequately identifying the measurement techniques or describing the quality of material used. The purpose of this document is to make available, in a single reference, an annotated bibliography and sets of properties for nine of the more attractive isotopes available for use in power production. Part I contains all the unclassified information that was available in the literature surveyed. Part II is the classified counterpart to Part I. -

United States Pmao" ICC Patented June 30, '1959 1 2 Adsorption on Manganese Dioxide, but It May on the 2,892,677 Other Hand He Left in the Solution
r. 2,892,677 United States Pmao" ICC Patented June 30, '1959 1 2 adsorption on manganese dioxide, but it may on the 2,892,677 other hand he left in the solution. If it is removed, a certain amount of manganese nitrate is formed from the ' SEPARATION OF URANIUM FROM THORIUM manganese dioxide in the solution and this nitrate reacts AND PROTACTINIUM with the sodium diethyldithiocarbamate to give manga William Kenneth Rodgerson Musgrave, Durham, Eng ' nese diethyldithiocarbamate, which must subsequently be land, assignor, by mesne assignments, to the United separated from the uranium. ,, . .States of America as represented by the United States Whether or not the protactinium has been removed, Atomic Energy Commission the solution is brought to a pH of between 2 and 3, for No Drawing. Application November 27, 1946 10 example by the addition of ammonia. The sodium di Serial No. 712,722 ethyldithiocarbamate is then dissolved in a solvent in which the subsequently formed uranyl diethyldithiocar '8 Claims. (Cl. 23—14.5) bamate is also soluble. Such a solvent is, for example, amyl acetate or methyl isobutyl ketone, the former being This invention relates to the separation of uranium 15 preferable for the reasons which will be indicated below. from thorium and protactinium. A mixture of these The sodium diethyldithiocarbamate is dissolved in amyl elements is obtained, for example, as the result of irradia acetate to form a solution containing 0.25% of the tion by neutrons of so-called thorium carbonate, which former, and this solution is then shaken up with the is a mixture of thorium oxide and thorium carbonate. -

Solvent Extraction of Uranium with Acetylacetone and Tri-N-Butyl
DAEHAN HWAHAK HWOEJEE (Journal of the Korean Chemical Society) Vol. 24, No. 3, 1980 Printed in the Republic of Korea 아세 틸아세톤과 트리부틸인산의 도데칸용액 에 의한 우라늄의 용매추출 裴奎善-鄰奇碩 韓國核燃料開發公團 (1980. 2. 27 接受 ) Solvent Extraction of Uranium with Acetylacetone and Tri-n-Butyl Phosphate in “-Dodecane Kyu Sun Bai and Key Suck Jung Korea Nuclear Fuel Development Institute, Daeduck 300-32, Korea (Received Feb. 27, 1980) 요 악 . 도데칸에 녹인 아세틸아세톤과 트리부틸인산으로 묽은 질산우라닐수용액에서 우라늄 (VI) 을 추출했다 . pHl 이상에서 이 혼합추출제의 상승적 효과가 관측되었다 . 추출되는 화학종은 1:2:1 및 1:2:2 우라닐 -아세털 아세톤 -트리부털 인산착물이다 . 이들 반응의 추출정수들을 측정하였다 . ABSTRACT. Uranium (VI) was extracted from dilute aqueous solutions of uranyl nitrate with acetylacetone and tri-n-butyl phosphate in n-dodecane. Synergistic effect was observed with the mixed reagents above pH 1, The species extracted are the 1:2:1 and the 1:2:2 uranyl-AA-TBP complexes. The extraction constants for these reactions have been determined. by crystallization of the salt as UO2(NO3)2 INTRODUCTION •6H2O. HAA and TBP were dissolved in do Acetylacetone (HAA) is a simple -diketone. decane and uranyl nitrate in water. Equal A weak acid with pKa=8. 2% HAA has long volumes of organic and aqueous phases were been used as a chelating agent for many equilibrated in 100 mZ separatory funnels by metals.2 It is quite soluble in water and its vigorous shaking for several minutes. Equili brium temperature was 25 °C or ambient. pH distribution coefficient is E=5. 95 for benzene/ water system.3 Because of its solubility in measurements were made by using a Corning water, HAA has not been so widely used as, Model 130 pH meter. -

Removal of Plutonium from Plutonium Hexafluoride--Uranium Hexafluoride
United States Patent n?) [in 3,708,568 Golliher et al. [45] Jan. 2,1973 [54] REMOVAL OF PLUTONIUM FROM 2,843,453 7/1958 Connicketal. .23/332 PLUTONIUM HEXAFLUORIDE- 3,165,376 1/1965 Golliher.. .23/337 URANIUM HEXAFLUORIDE 3,178,258 4/1965 Cathersetal 23/337 MIXTURES 3,423,190 1/1969 Steindler et al .23/326 [75] Inventors: Waldo R. Golliher; Robert L. Har- Primary Examiner—Carl D. Quarforth ris; Reynold A. LeDoux, Jr., all ol Assistant Examiner—F. M. Gittes Paducah, Ky. Attorney—Roland A. Anderson [73] Assignee: The United States of America as represented by the United State: [57] ABSTRACT Atomic Energy Commission This invention relates to a method of selectively [22] Filed: Oct. 20,1970 removing plutonium values from a fluid mixture con- [21] Appl. No.: 82,508 taining plutonium hexafluoride and uranium hex- afluoride by passing the mixture through a bed of pel- [52] U.S. C! 423/6,423/19, 252/301.1 R letized cobaltous fluoride at a temperature in the [51] Int. CI COlg 56/00 range 134° to 1,000° F. to effect removal of plutonium [58] Field of Search 55/74; 252/301.1 R; 23/332, by the cobaltous fluoride. 23/326, 337, 344, 352; 423/19, 6, 11, 251, 258 [56] References Cited 3 Claims, No Drawings UNITED STATES PATENTS 3,615,267 10/1971 Golliher et al. 23/343 3,725,661 3 4 REMOVAL OF PLUTONIUM FROM PLUTONIUM recovered. The cobaltous fluoride can then be HEXAFLUORIDE-URANIUM HEXAFLUORIDE processed for reuse by contacting with gaseous MIXTURES hydrogen at a temperature in the range 400° to 500° F. -

System Studies of Fission-Fusion Hybrid Molten Salt Reactors
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2013 SYSTEM STUDIES OF FISSION-FUSION HYBRID MOLTEN SALT REACTORS Robert D. Woolley University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Nuclear Engineering Commons Recommended Citation Woolley, Robert D., "SYSTEM STUDIES OF FISSION-FUSION HYBRID MOLTEN SALT REACTORS. " PhD diss., University of Tennessee, 2013. https://trace.tennessee.edu/utk_graddiss/2628 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Robert D. Woolley entitled "SYSTEM STUDIES OF FISSION-FUSION HYBRID MOLTEN SALT REACTORS." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Nuclear Engineering. Laurence F. Miller, Major Professor We have read this dissertation and recommend its acceptance: Ronald E. Pevey, Arthur E. Ruggles, Robert M. Counce Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) SYSTEM STUDIES OF FISSION-FUSION HYBRID MOLTEN SALT REACTORS A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Robert D. -

RFI) DE-FOA-0002533 on Cleaning up Radioisotope Enventories (CURIE)
U.S. Department of Energy Advanced Research Projects Agency – Energy (ARPA-E) Request for Information (RFI) DE-FOA-0002533 on Cleaning Up RadioIsotope Enventories (CURIE) Introduction The purpose of this RFI is to solicit input for a potential future ARPA-E research program focused on the development of technologies that would enable the effective management of the Nation’s commercial used nuclear fuel (UNF). The goals of this RFI are to (1) solicit information about reactor fuel needs for both the current commercial light-water reactor (LWR) fleet and future advanced reactors, and (2) seek insights into technology gaps and/or cost drivers that may be hindering economical recycling of existing LWR UNF.1 This information is needed to help ARPA-E identify ways in which the Nation’s roughly 86,000 MTU2 inventory of UNF, which has been increasing by approximately 2,000 MTU per year, can best be recycled to support current and advanced reactor fuel needs. Such activities are consistent with ARPA-E’s statutory goals, which include supporting the development of transformative solutions for addressing UNF.3 ARPA-E is interested in information about technologies with the potential to make recycling UNF at least as economical, safe, and secure as the current once-through fuel cycle.4 Such technologies would enable a UNF treatment facility to be economically constructed, managed, and operated; yield an actinide product that is cost-competitive with natural uranium (U) obtained from traditional mining and milling; and generate significantly lower -

Material Safety Data Sheet
SAFETY DATA SHEET URANYL NITRATE SOLUTION SECTION 1: CHEMICAL PRODUCT & COMPANY IDENTIFICATION New Brunswick Laboratory U.S. Department of Energy 9800 South Cass Avenue Argonne, IL 60439 1-630-252-CRMS (2767) Emergency Phone Numbers: 1-630-252-6130 or 1-630-252-5731 Chemical Name: Uranyl Nitrate Solution, UO2(NO3)2 Other Identifiers: Certified Reference Material (CRM) standard or Safeguards Measurement Evaluation (SME) sample Use and Restrictions: This material is prepared for use as a standard or in inter-laboratory comparison programs at analytical laboratories, which routinely handle uranium and/or plutonium. NBL expects that recipients of their material are in compliance with 29 CFR 1910.1200(h) which requires employers to provide employees with effective information and training on hazardous chemicals in their work area. SECTION 2: HAZARDS IDENTIFICATION OSHA HAZARDS: Skin Corrosion/Irritation Category 1B. Serious Eye Damage/Irritation Category 1 TARGET ORGANS: Skin, Eyes, Kidneys. GHS Label elements, including precautionary statements Signal Word: DANGER Pictogram: Page 1 of 12 Hazard Statement(s) H314 Causes severe skin burns and eye damage H373 May cause damage to organs through prolonged or repeated exposure. H411 Toxic to aquatic life with long lasting effects. Precautionary statement(s) P260 Do not breathe dust/ fume/ gas/ mist/ vapors/ spray. P262 Do not get in eyes, on skin, or on clothing. P264 Wash skin thoroughly after handling. P273 Avoid release to the environment. P280 Wear protective gloves/protective clothing/eye protection/face protection P310 Immediately call a POISON CENTER or doctor/ physician if swallowed or inhaled. Other Hazard(s): Radioactive NFPA RATINGS (SCALE 0-4): Health=3 Fire=0 Reactivity=0 Special Hazard= OX SECTION 3: COMPOSITION/INFORMATION ON INGREDIENTS Chemical Name: Uranyl Nitrate Solution Common Names/Synonyms: CRM U045; CRM 135; CRM 145; CRM 145-B; CRM U930-D; Safeguards Measurement Evaluation (SME) Low Enrichment, Normal Enrichment, or High Enrichment Solutions; Uranyl Nitrate in Nitric Acid Solution. -

Export Control Handbook for Chemicals
Export Control Handbook for Chemicals -Dual-use control list -Common Military List -Explosives precursors -Syria restrictive list -Psychotropics and narcotics precursors ARNES-NOVAU, X 2019 EUR 29879 This publication is a Technical report by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide evidence-based scientific support to the European policymaking process. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Xavier Arnés-Novau Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-785421 Filippo Sevini Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-786793 EU Science Hub https://ec.europa.eu/jrc JRC 117839 EUR 29879 Print ISBN 978-92-76-11971-5 ISSN 1018-5593 doi:10.2760/844026 PDF ISBN 978-92-76-11970-8 ISSN 1831-9424 doi:10.2760/339232 Luxembourg: Publications Office of the European Union, 2019 © European Atomic Energy Community, 2019 The reuse policy of the European Commission is implemented by Commission Decision 2011/833/EU of 12 December 2011 on the reuse of Commission documents (OJ L 330, 14.12.2011, p. 39). Reuse is authorised, provided the source of the document is acknowledged and its original meaning or message is not distorted. The European Commission shall not be liable for any consequence stemming from the reuse. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals