Review of Uranyl Mineral Solubility Measurements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction Processing of Concentrated Solutions of Uranyl Nitrate with High Impurities Content V.M
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Electronic archive of Tomsk Polytechnic University Сhemistry UDC 546.791.02.238:66.061.51 EXTRACTION PROCESSING OF CONCENTRATED SOLUTIONS OF URANYL NITRATE WITH HIGH IMPURITIES CONTENT V.M. Korotkevich, V.V.Lazarchuck, T.G. Shikerun, V.I. Shamin, N.A. Mikhailova, F.A. Dorda Federal Unitary Enterprise Siberian Group of Chemical Enterprises, Seversk Еmail: [email protected] Process flowsheet of recycling uranium concentrated solutions with its purification from insoluble impurities of iron, silicon, molybde num, calcium oxides and hydroxides and soluble impurities with application of centrifugal extractors cascade has been developed and suggested for commercial introduction. The process was carried out at extractant saturation (30 % tributyl phosphate in hydrocarbon diluent) in extraction assembly lower than a limiting level (85...95 g/l) and in wash assembly – at limiting saturation (up to 120 g/l). As a result the waste uranium content in watertail solutions 0,01...0,04 g/l and minimal content of impurities in reextractors is provided. Perspective program on development of nuclear technology are also: reliability of hermetization, possi power engineering proposes to involve wide range of raw bility of remote service, resistance to corrosive attack of materials from uranium ores to secondary uranium ma structural materials, duration of overhaul period etc. terials into processing at atomic enterprises. In this case In Russia the centrifugal extractor of ECТseries the problem of reprocessing concentrated uranium so with continuous extraction of solid phase [3], which may lutions with high impurity content occurs. -

Françoisite-(Ce)
American Mineralogist, Volume 95, pages 1527–1532, 2010 Françoisite-(Ce), a new mineral species from La Creusaz uranium deposit (Valais, Switzerland) and from Radium Ridge (Flinders Ranges, South Australia): Description and genesis NICOLAS MEISSER,1,* JOËL BRUGGER,2,3 STEFA N AN SER M ET ,1 PHILI pp E THÉLI N ,4 A N D FRA N ÇOIS BUSSY 5 1Musée de Géologie and Laboratoire des Rayons-X, Institut de Minéralogie et de Géochimie, UNIL, Anthropole, CH-1015 Lausanne-Dorigny, Switzerland 2South Australian Museum, North Terrace, 5000 Adelaide, Australia 3TRaX, School of Earth and Environmental Sciences, University of Adelaide, 5005 Adelaide, Australia 4Laboratoire des Rayons-X, Institut de Minéralogie et de Géochimie, UNIL, Anthropole, CH-1015 Lausanne-Dorigny, Switzerland 5Laboratoire de la microsonde électronique, Institut de Minéralogie et de Géochimie, UNIL, Anthropole, CH-1015 Lausanne-Dorigny, Switzerland AB STRACT The new mineral françoisite-(Ce), (Ce,Nd,Ca)[(UO2)3O(OH)(PO4)2]·6H2O is the Ce-analog of françoisite-(Nd). It has been discovered simultaneously at the La Creusaz uranium deposit near Les Marécottes in Valais, Switzerland, and at the Number 2 uranium Workings, Radium Ridge near Mt. Painter, Arkaroola area, Northern Flinders Ranges in South Australia. Françoisite-(Ce) is a uranyl- bearing supergene mineral that results from the alteration under oxidative conditions of REE- and U4+- bearing hypogene minerals: allanite-(Ce), monazite-(Ce), ±uraninite at Les Marécottes; monazite-(Ce), ishikawaite-samarskite, and an unknown primary U-mineral at Radium Ridge. The REE composition of françoisite-(Ce) results from a short aqueous transport of REE leached out of primary minerals [most likely monazite-(Ce) at Radium Ridge and allanite-(Ce) at La Creusaz], with fractionation among REE resulting mainly from aqueous transport, with only limited Ce loss due to oxidation to Ce4+ during transport. -

A Protein Engineered to Bind Uranyl Selectively and with Femtomolar Affinity
ARTICLES PUBLISHED ONLINE: 26 JANUARY 2014 | DOI: 10.1038/NCHEM.1856 A protein engineered to bind uranyl selectively and with femtomolar affinity Lu Zhou1†, Mike Bosscher1†, Changsheng Zhang2,3†,SalihO¨ zc¸ubukc¸u1, Liang Zhang1, Wen Zhang1, Charles J. Li1, Jianzhao Liu1, Mark P. Jensen4, Luhua Lai2,3* and Chuan He1* 21 ∼ Uranyl (UO2 ), the predominant aerobic form of uranium, is present in the ocean at a concentration of 3.2 parts per 109 (13.7 nM); however, the successful enrichment of uranyl from this vast resource has been limited by the high concentrations of metal ions of similar size and charge, which makes it difficult to design a binding motif that is selective for uranyl. Here we report the design and rational development of a uranyl-binding protein using a computational screening process in the initial search for potential uranyl-binding sites. The engineered protein is thermally stable and offers very high affinity and selectivity for uranyl with a Kd of 7.4 femtomolar (fM) and >10,000-fold selectivity over other metal ions. We also demonstrated that the uranyl-binding protein can repeatedly sequester 30–60% of the uranyl in synthetic sea water. The chemical strategy employed here may be applied to engineer other selective metal-binding proteins for biotechnology and remediation applications. ranium is the key element for nuclear-energy production and Through billions of years of evolution nature has produced is important in many other applications. The most stable and strategies to recognize beneficial or toxic metal ions with high Urelevant uranium ion in aerobic environments is the uranyl sensitivity and selectivity. -

Hierarchically-Structured Metalloprotein Composite
ARTICLE There are amendments to this paper https://doi.org/10.1038/s41467-020-14709-y OPEN Hierarchically-structured metalloprotein composite coatings biofabricated from co-existing condensed liquid phases ✉ Franziska Jehle 1, Elena Macías-Sánchez1, Sanja Sviben1, Peter Fratzl1, Luca Bertinetti 1 & ✉ Matthew J. Harrington 1,2 1234567890():,; Complex hierarchical structure governs emergent properties in biopolymeric materials; yet, the material processing involved remains poorly understood. Here, we investigated the multi- scale structure and composition of the mussel byssus cuticle before, during and after for- mation to gain insight into the processing of this hard, yet extensible metal cross-linked protein composite. Our findings reveal that the granular substructure crucial to the cuticle’s function as a wear-resistant coating of an extensible polymer fiber is pre-organized in con- densed liquid phase secretory vesicles. These are phase-separated into DOPA-rich proto- granules enveloped in a sulfur-rich proto-matrix which fuses during secretion, forming the sub-structure of the cuticle. Metal ions are added subsequently in a site-specific way, with iron contained in the sulfur-rich matrix and vanadium coordinated by DOPA-catechol in the granule. We posit that this hierarchical structure self-organizes via phase separation of specific amphiphilic proteins within secretory vesicles, resulting in a meso-scale structuring that governs cuticle function. 1 Department of Biomaterials, Max Planck Institute of Colloids and Interfaces, 14424 Potsdam, -

Depleted Uranium Technical Brief
Disclaimer - For assistance accessing this document or additional information,please contact [email protected]. Depleted Uranium Technical Brief United States Office of Air and Radiation EPA-402-R-06-011 Environmental Protection Agency Washington, DC 20460 December 2006 Depleted Uranium Technical Brief EPA 402-R-06-011 December 2006 Project Officer Brian Littleton U.S. Environmental Protection Agency Office of Radiation and Indoor Air Radiation Protection Division ii iii FOREWARD The Depleted Uranium Technical Brief is designed to convey available information and knowledge about depleted uranium to EPA Remedial Project Managers, On-Scene Coordinators, contractors, and other Agency managers involved with the remediation of sites contaminated with this material. It addresses relative questions regarding the chemical and radiological health concerns involved with depleted uranium in the environment. This technical brief was developed to address the common misconception that depleted uranium represents only a radiological health hazard. It provides accepted data and references to additional sources for both the radiological and chemical characteristics, health risk as well as references for both the monitoring and measurement and applicable treatment techniques for depleted uranium. Please Note: This document has been changed from the original publication dated December 2006. This version corrects references in Appendix 1 that improperly identified the content of Appendix 3 and Appendix 4. The document also clarifies the content of Appendix 4. iv Acknowledgments This technical bulletin is based, in part, on an engineering bulletin that was prepared by the U.S. Environmental Protection Agency, Office of Radiation and Indoor Air (ORIA), with the assistance of Trinity Engineering Associates, Inc. -
![A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4n]3[UO2(Ncse)5]](https://docslib.b-cdn.net/cover/7628/a-structural-and-spectroscopic-study-of-the-first-uranyl-selenocyanate-et4n-3-uo2-ncse-5-307628.webp)
A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4n]3[UO2(Ncse)5]
inorganics Article A Structural and Spectroscopic Study of the First Uranyl Selenocyanate, [Et4N]3[UO2(NCSe)5] Stefano Nuzzo, Michelle P. Browne, Brendan Twamley, Michael E. G. Lyons and Robert J. Baker * School of Chemistry, Trinity College, University of Dublin, 2 Dublin, Ireland; [email protected] (S.N.); [email protected] (M.P.B.); [email protected] (B.T.); [email protected] (M.E.G.L.) * Correspondence: [email protected]; Tel.: +353-1-896-3501; Fax: +353-1-671-2826 Academic Editors: Stephen Mansell and Steve Liddle Received: 30 October 2015; Accepted: 4 February 2016; Published: 16 February 2016 Abstract: The first example of a uranyl selenocyanate compound is reported. The compound [Et4N]3[UO2(NCSe)5] has been synthesized and fully characterized by vibrational and multinuclear (1H, 13C{1H} and 77Se{1H}) NMR spectroscopy. The photophysical properties have also been recorded and trends in a series of uranyl pseudohalides discussed. Spectroscopic evidence shows that the U–NCSe bonding is principally ionic. An electrochemical study revealed that the reduced uranyl(V) species is unstable to disproportionation and a ligand based oxidation is also observed. The structure of [Et4N]4[UO2(NCSe)5][NCSe] is also presented and Se¨ ¨ ¨ H–C hydrogen bonding and Se¨ ¨ ¨ Se chalcogen–chalcogen interactions are seen. Keywords: uranyl; structural determination; photophysics 1. Introduction The chemistry of uranium in its highest oxidation state has held scientists fascination for a long 2+ period of time. The uranyl moiety, [UO2] , is well studied in aqueous phases due, in part, to relevance in the nuclear waste treatment. Moreover, the photophysical properties of uranyl were first used in ancient roman times in colored glass [1], whilst comprehensive understanding of the bonding, and therefore photophysical properties, has come from both experiment and theory. -

Weeksite K2(UO2)2(Si5o13)·4H2O
Weeksite K2(UO2)2(Si5O13)·4H2O Crystal Data: Monoclinic. Point Group: 2/m: As bladed or acicular crystals, flattened on {010} and elongated along [001], and as flat plates; also as spherulites and radiating fibrous clusters. - Twinning: By two-fold rotation around [401 ]. Physical Properties: Cleavage: Two good prismatic cleavages noted. Hardness = < 2 D(meas.) = ~ 4.1 D(calc.) = 3.80 Radioactive. Optical Properties: Transparent to translucent. Color: Yellow. Luster: Waxy to silky. Optical Class: Biaxial (-). α = 1.596 β = 1.603 γ = 1.606 2V(meas.) = ~ 60° 2V(calc.) = 66° Pleochroism: X = colorless, Y = pale yellow-green, Z = yellow-green. Dispersion: r > v, strong. Orientation: X = b, Y = c, Z = a. Cell Data: Space Group: C2/m. a = 14.26(2) b = 35.88(10) c = 14.20(2) β = 111.578(3)° Z = 4 X-ray Powder Pattern: Thomas Range, Utah, USA. 7.11 (10), 5.57 (9), 8.98 (8), 3.55 (7), 3.30 (7), 2.91 (6), 3.20 (5) Chemistry: (1) (1) Na2O 0.53 Al2O3 0.6 K2O 4.73 SiO2 29.44 CaO 0.67 UO3 55.78 BaO 3.11 H2O [7.02] MgO 0.18 Total 101.28 SrO 0.20 (1) Anderson mine, Arizona, USA; average of 8 electron microprobe analyses, supplemented by TGA, H2O calculated from structure, corresponds to (K1.031Na0.176Ca0.123Ba0.208)Σ=1.537(UO2)2.002 (Si5.030O13)·4H2O. Occurrence: In “opal" veinlets in rhyolite, agglomerates, sandstones and limestones. Association: “Opal," “chalcedony," calcite, gypsum, fluorite, uraninite, thorogummite, uranophane, boltwoodite, carnotite, margaritasite. Distribution: In the USA, in Utah, at the Autunite No. -
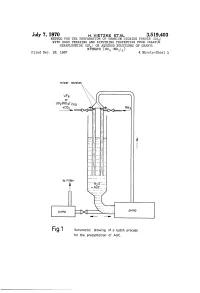
Nitrate (UO2 (NO)) 4
July 7, 1970 H. VETZKE ETA 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Fied Dec. 18, 1967 NiTRATE (UO2 (NO)) 4. Sheets-Sheet l mixer nozzles --race to fitter Fig.1 Schematic drawing of a batch process for the precipitation of AUC. July 7, 1970 H. VETZKE EA 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM RANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Filed Dec. 8, 1967 NiTRATE (UO2 (NO)) 4. Sheets-Sheet 3. mixer nozzles UF6 Of U02 (NO3)2ad tC92--X Ex-Ns / to filter cooling precipitation Vessel vessel Fig. 2 Schematic drawing of a continuous process for the precipitation of AUC July 7, 1970 H, VETZKE ETAL 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL Filed Dec. 18, 1967 NiTRATE (UO (NO)2) 4. Sheets-Sheet 3 Off gas Off gas 487\m Nx UO2 powder XXX.& X.X S& steam / H2 Fig. 3 Ot Schematic drawing of a fluidized bed furnace for the reduction of AUC to U02, July 7, 1970 H. VEZKE ETAL 3,519,403 METHOD FOR THE PREPARATION OF URANIUM DIOXIDE POWDER (UO) WITH GOOD PRESSING AND SINTERING PROPERTIES FROM URANIUM HEXAFLUORIDE (UF) OR AQUEOUS SOLUTIONS OF URANYL NITRATE (UO2 (NO)a) Filed Dec. 18, 1967 4. Sheets-Sheet 1 -- Offgas Step 1: Decomposition, reduction and pyrohydrolysis i Step 2: Pyrohydrolysis Step 3: Controlled oxidation an o- are as an un- Fig. -

Procedure for Determining Uranium, Plutonium and Americium by Extraction-Chromatographic Procedures
Procedure for determining uranium, plutonium and americium by extraction-chromatographic procedures H-U/Pu/Am-AWASS-01 Authors: M. Beyermann D. Obrikat Federal coordinating office for drinking water, groundwater, wastewater, sludge, waste and wastewater of nuclear power plants (Leitstelle für die Überwachung der Radioaktivität in Trinkwasser, Grundwasser, Abwasser, Klärschlamm, Reststoffen und Abfällen) ISSN 1865-8725 Version October 2000 Procedures manual for monitoring of radioactive substances in the environment and of external radiation (Messanleitungen für die „Überwachung radioaktiver Stoffe in der Umwelt und externer Strahlung“) H-U/Pu/Am-AWASS-01-01 Procedure for determining uranium, plutonium and americium by means of extraction- chromatographic procedures 1 Scope This procedure serves to simultaneously determine the uranium isotopes U-234, U-235 and U-238, the plutonium isotopes Pu-238, Pu-239 and Pu-240, as well as the americium isotope Am-241 in samples of wastewater from nuclear facilities. It furthermore offers an option of determining the curium isotopes Cm-242 and Cm-244 without further effort. For determining Pu-241, reference is made to pro- cedure H-Pu-241-AWASS-01 of these measuring instructions. 2 Sampling As far as sampling is concerned, reference is made to procedure H-SPEKT- AWASS-01 of these measuring instructions. The sample of the wastewater to be analysed is acidified with ca. 10 ml of concentrated nitric acid (14 mol·l-1) per litre to a pH of about 1. The stability of the acidic reaction needs to be monitored, in particular if the sample is stored for an extended period of time. This procedure ensures that a detection limit of 0,05 Bq·l-1 for alpha-emitters is reached in a sample volume of 0,1 litres to 0,25 litres and thus complies with the nuclear safety standard 1504 of the Nuclear Safety Standards Commission (1). -

United States Pmao" ICC Patented June 30, '1959 1 2 Adsorption on Manganese Dioxide, but It May on the 2,892,677 Other Hand He Left in the Solution
r. 2,892,677 United States Pmao" ICC Patented June 30, '1959 1 2 adsorption on manganese dioxide, but it may on the 2,892,677 other hand he left in the solution. If it is removed, a certain amount of manganese nitrate is formed from the ' SEPARATION OF URANIUM FROM THORIUM manganese dioxide in the solution and this nitrate reacts AND PROTACTINIUM with the sodium diethyldithiocarbamate to give manga William Kenneth Rodgerson Musgrave, Durham, Eng ' nese diethyldithiocarbamate, which must subsequently be land, assignor, by mesne assignments, to the United separated from the uranium. ,, . .States of America as represented by the United States Whether or not the protactinium has been removed, Atomic Energy Commission the solution is brought to a pH of between 2 and 3, for No Drawing. Application November 27, 1946 10 example by the addition of ammonia. The sodium di Serial No. 712,722 ethyldithiocarbamate is then dissolved in a solvent in which the subsequently formed uranyl diethyldithiocar '8 Claims. (Cl. 23—14.5) bamate is also soluble. Such a solvent is, for example, amyl acetate or methyl isobutyl ketone, the former being This invention relates to the separation of uranium 15 preferable for the reasons which will be indicated below. from thorium and protactinium. A mixture of these The sodium diethyldithiocarbamate is dissolved in amyl elements is obtained, for example, as the result of irradia acetate to form a solution containing 0.25% of the tion by neutrons of so-called thorium carbonate, which former, and this solution is then shaken up with the is a mixture of thorium oxide and thorium carbonate. -

Solvent Extraction of Uranium with Acetylacetone and Tri-N-Butyl
DAEHAN HWAHAK HWOEJEE (Journal of the Korean Chemical Society) Vol. 24, No. 3, 1980 Printed in the Republic of Korea 아세 틸아세톤과 트리부틸인산의 도데칸용액 에 의한 우라늄의 용매추출 裴奎善-鄰奇碩 韓國核燃料開發公團 (1980. 2. 27 接受 ) Solvent Extraction of Uranium with Acetylacetone and Tri-n-Butyl Phosphate in “-Dodecane Kyu Sun Bai and Key Suck Jung Korea Nuclear Fuel Development Institute, Daeduck 300-32, Korea (Received Feb. 27, 1980) 요 악 . 도데칸에 녹인 아세틸아세톤과 트리부틸인산으로 묽은 질산우라닐수용액에서 우라늄 (VI) 을 추출했다 . pHl 이상에서 이 혼합추출제의 상승적 효과가 관측되었다 . 추출되는 화학종은 1:2:1 및 1:2:2 우라닐 -아세털 아세톤 -트리부털 인산착물이다 . 이들 반응의 추출정수들을 측정하였다 . ABSTRACT. Uranium (VI) was extracted from dilute aqueous solutions of uranyl nitrate with acetylacetone and tri-n-butyl phosphate in n-dodecane. Synergistic effect was observed with the mixed reagents above pH 1, The species extracted are the 1:2:1 and the 1:2:2 uranyl-AA-TBP complexes. The extraction constants for these reactions have been determined. by crystallization of the salt as UO2(NO3)2 INTRODUCTION •6H2O. HAA and TBP were dissolved in do Acetylacetone (HAA) is a simple -diketone. decane and uranyl nitrate in water. Equal A weak acid with pKa=8. 2% HAA has long volumes of organic and aqueous phases were been used as a chelating agent for many equilibrated in 100 mZ separatory funnels by metals.2 It is quite soluble in water and its vigorous shaking for several minutes. Equili brium temperature was 25 °C or ambient. pH distribution coefficient is E=5. 95 for benzene/ water system.3 Because of its solubility in measurements were made by using a Corning water, HAA has not been so widely used as, Model 130 pH meter. -

CNWRA Paper Element Mobility in Uranium Deposits of the Sierra
Elemental Mobility in Uranium Deposits of the Sierra Peiia Blanca as a Natural Analog of Radionuclide Migration in a High-Level Nuclear Waste Repository English C. Pearcy and M. Murphy Southwest ResearchWilliam Institute San Antonio, Texas Phillip C. Goodell University of Texas El Paso, Texas For many years research interest in uranium ore deposits was mainly directed toward understanding the mechanisms and processes of formation of the deposits. Motivation for these studies derived from the economic value of the uranium as fuel for nuclear reactors. More recently, concern with disposal of the spent nuclear fuel has prompted research on uranium deposits because they can provide information on the behavior of nuclear waste in geologic repositories. Because high-level nuclear waste (HLW) will constitute a threat to public safety for many thousands of years, it is necessary to attempt to predict the performance of a HLW repository far into the future. Although such times are short on a geologic scale, they are far in excess of experimental capabilities. In addition, the size of a geologic repository greatly exceed that of laboratory facilities. One approach to theseHLW problems of large timewill and space scales is to study natural systems. For example, natural materials which approximate components of a repository may be studied to gain a better understanding of the behavior to be expected from the repsirory marerial. Similarly, a process which occurs (or has occurred) in nature and which may be significant to the performance of a repository may be investigated to learn of possible effects on a repository system. By studying such natural analogs, which have existed for time spans comparable to radionuclide isolation requirements and which are of comparable scale to a repository, the uncertainty inherent in projecting such large scale processes far into the future can be reduced.