Downloaded from the NCBI Website (Table S1)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SALT TOLERANT PLANTS Recommended for Pender County Landscapes
North Carolina Cooperative Extension NC STATE UNIVERSITY SALT TOLERANT PLANTS Recommended for Pender County Landscapes Pender County Cooperative Extension Urban Horticulture Leaflet 14 Coastal Challenges Plants growing at the beach are subjected to environmental conditions much different than those planted further inland. Factors such as blowing sand, poor soils, high temperatures, and excessive drainage all influence how well plants perform in coastal landscapes, though the most significant effect on growth is salt spray. Most plants will not tolerate salt accumulating on their foliage, making plant selection for beachfront land- scapes particularly challenging. Salt Spray Salt spray is created when waves break on the beach, throwing tiny droplets of salty water into the air. On-shore breezes blow this salt laden air landward where it comes in contact with plant foliage. The amount of salt spray plants receive varies depending on their proximity to the beachfront, creating different vegetation zones as one gets further away from the beachfront. The most salt-tolerant species surviving in the frontal dune area. As distance away from the ocean increases, the level of salt spray decreases, allowing plants with less salt tolerance to survive. Natural Protection The impact of salt spray on plants can be lessened by physically blocking salt laden winds. This occurs naturally in the maritime forest, where beachfront plants protect landward species by creating a layer of foliage that blocks salt spray. It is easy to see this effect on the ocean side of maritime forest plants, which are “sheared” by salt spray, causing them to grow at a slant away from the oceanfront. -

Conservation Assessment for the Bigleaf Snowbell (Styrax Grandifolius Ait.)
Conservation Assessment for the Bigleaf Snowbell (Styrax grandifolius Ait.) Steven R. Hill, Ph.D. Division of Biodiversity and Ecological Entomology Biotic Surveys and Monitoring Section 1816 South Oak Street Champaign, Illinois 61820 Prepared for the U.S.D.A. Forest Service, Eastern Region (Region 9), Shawnee and Hoosier National Forests INHS Technical Report 2007 (65) Date of Issue: 17 December 2007 Cover photo: Styrax grandifolius Ait., from the website: In Bloom – A Monthly Record of Plants in Alabama; Landscape Horticulture at Auburn University, Auburn, Alabama. http://www.ag.auburn.edu/hort/landscape/inbloomapril99.html This Conservation Assessment was prepared to compile the published and unpublished information on the subject taxon or community; or this document was prepared by another organization and provides information to serve as a Conservation Assessment for the Eastern Region of the Forest Service. It does not represent a management decision by the U.S. Forest Service. Though the best scientific information available was used and subject experts were consulted in preparation of this document, it is expected that new information will arise. In the spirit of continuous learning and adaptive management, if you have information that will assist in conserving the subject taxon, please contact the Eastern Region of the Forest Service - Threatened and Endangered Species Program at 310 Wisconsin Avenue, Suite 580 Milwaukee, Wisconsin 53203. 2 Conservation Assessment for the Bigleaf Snowbell (Styrax grandifolius Ait.) Table of Contents -

Ocn319079567-2020-09-04.Pdf (471.0Kb)
Visit The University of Massachusetts Amherst Apply Give Search UMass.edu (/) Coronavirus (COVID-19) Resources from UMass Extension and the Center for Agriculture, Food and the Environment: ag.umass.edu/coronavirus (/coronavirus) LNUF Home (/landscape) About (/landscape/about) Newsletters & Updates (/landscape/newsletters-updates) Publications & Resources (/landscape/publications-resources) Services (/landscape/services) Education & Events (/landscape/upcoming-events) Make a Gift (https://securelb.imodules.com/s/1640/alumni/index.aspx?sid=1640&gid=2&pgid=443&cid=1121&dids=2540) Landscape Message: September 4, 2020 September 4, 2020 Issue: 16 UMass Extension's Landscape Message is an educational newsletter intended to inform and guide Massachusetts Green Industry professionals in the management of our collective landscape. (/landscape) Detailed reports from scouts and Extension specialists on growing conditions, pest activity, and Search CAFE cultural practices for the management of woody ornamentals, trees, and turf are regular features. The following issue has been updated to provide timely management information and the latest Search this site regional news and environmental data. Search Registration has begun for our UMass Extension GREEN SCHOOL! (/sites/ag.umass.edu/files/pest- alerts/images/content/green_school_2020_virtual.jpg) Green Newsletters & School is going VIRTUAL for 2020! Classes will be Oct. 26 - Dec. Updates 10. This comprehensive 12-day certificate short course for Green Landscape Message Industry professionals is taught -

Cally Plant List a ACIPHYLLA Horrida
Cally Plant List A ACIPHYLLA horrida ACONITUM albo-violaceum albiflorum ABELIOPHYLLUM distichum ACONITUM cultivar ABUTILON vitifolium ‘Album’ ACONITUM pubiceps ‘Blue Form’ ACAENA magellanica ACONITUM pubiceps ‘White Form’ ACAENA species ACONITUM ‘Spark’s Variety’ ACAENA microphylla ‘Kupferteppich’ ACONITUM cammarum ‘Bicolor’ ACANTHUS mollis Latifolius ACONITUM cammarum ‘Franz Marc’ ACANTHUS spinosus Spinosissimus ACONITUM lycoctonum vulparia ACANTHUS ‘Summer Beauty’ ACONITUM variegatum ACANTHUS dioscoridis perringii ACONITUM alboviolaceum ACANTHUS dioscoridis ACONITUM lycoctonum neapolitanum ACANTHUS spinosus ACONITUM paniculatum ACANTHUS hungaricus ACONITUM species ex. China (Ron 291) ACANTHUS mollis ‘Long Spike’ ACONITUM japonicum ACANTHUS mollis free-flowering ACONITUM species Ex. Japan ACANTHUS mollis ‘Turkish Form’ ACONITUM episcopale ACANTHUS mollis ‘Hollard’s Gold’ ACONITUM ex. Russia ACANTHUS syriacus ACONITUM carmichaelii ‘Spätlese’ ACER japonicum ‘Aconitifolium’ ACONITUM yezoense ACER palmatum ‘Filigree’ ACONITUM carmichaelii ‘Barker’s Variety’ ACHILLEA grandifolia ACONITUM ‘Newry Blue’ ACHILLEA ptarmica ‘Perry’s White’ ACONITUM napellus ‘Bergfürst’ ACHILLEA clypeolata ACONITUM unciniatum ACIPHYLLA monroi ACONITUM napellus ‘Blue Valley’ ACIPHYLLA squarrosa ACONITUM lycoctonum ‘Russian Yellow’ ACIPHYLLA subflabellata ACONITUM japonicum subcuneatum ACONITUM meta-japonicum ADENOPHORA aurita ACONITUM napellus ‘Carneum’ ADIANTUM aleuticum ‘Japonicum’ ACONITUM arcuatum B&SWJ 774 ADIANTUM aleuticum ‘Miss Sharples’ ACORUS calamus ‘Argenteostriatus’ -

Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi
The University of Southern Mississippi The Aquila Digital Community Honors Theses Honors College Spring 5-2016 Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi Hanna M. Miller University of Southern Mississippi Follow this and additional works at: https://aquila.usm.edu/honors_theses Part of the Biodiversity Commons, and the Botany Commons Recommended Citation Miller, Hanna M., "Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi" (2016). Honors Theses. 389. https://aquila.usm.edu/honors_theses/389 This Honors College Thesis is brought to you for free and open access by the Honors College at The Aquila Digital Community. It has been accepted for inclusion in Honors Theses by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. The University of Southern Mississippi Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi by Hanna Miller A Thesis Submitted to the Honors College of The University of Southern Mississippi in Partial Fulfillment of the Requirement for the Degree of Bachelor of Science in the Department of Biological Sciences May 2016 ii Approved by _________________________________ Mac H. Alford, Ph.D., Thesis Adviser Professor of Biological Sciences _________________________________ Shiao Y. Wang, Ph.D., Chair Department of Biological Sciences _________________________________ Ellen Weinauer, Ph.D., Dean Honors College iii Abstract The North American Coastal Plain contains some of the highest plant diversity in the temperate world. However, most of the region has remained unstudied, resulting in a lack of knowledge about the unique plant communities present there. -

Outline of Angiosperm Phylogeny
Outline of angiosperm phylogeny: orders, families, and representative genera with emphasis on Oregon native plants Priscilla Spears December 2013 The following listing gives an introduction to the phylogenetic classification of the flowering plants that has emerged in recent decades, and which is based on nucleic acid sequences as well as morphological and developmental data. This listing emphasizes temperate families of the Northern Hemisphere and is meant as an overview with examples of Oregon native plants. It includes many exotic genera that are grown in Oregon as ornamentals plus other plants of interest worldwide. The genera that are Oregon natives are printed in a blue font. Genera that are exotics are shown in black, however genera in blue may also contain non-native species. Names separated by a slash are alternatives or else the nomenclature is in flux. When several genera have the same common name, the names are separated by commas. The order of the family names is from the linear listing of families in the APG III report. For further information, see the references on the last page. Basal Angiosperms (ANITA grade) Amborellales Amborellaceae, sole family, the earliest branch of flowering plants, a shrub native to New Caledonia – Amborella Nymphaeales Hydatellaceae – aquatics from Australasia, previously classified as a grass Cabombaceae (water shield – Brasenia, fanwort – Cabomba) Nymphaeaceae (water lilies – Nymphaea; pond lilies – Nuphar) Austrobaileyales Schisandraceae (wild sarsaparilla, star vine – Schisandra; Japanese -

Alphabetical Lists of the Vascular Plant Families with Their Phylogenetic
Colligo 2 (1) : 3-10 BOTANIQUE Alphabetical lists of the vascular plant families with their phylogenetic classification numbers Listes alphabétiques des familles de plantes vasculaires avec leurs numéros de classement phylogénétique FRÉDÉRIC DANET* *Mairie de Lyon, Espaces verts, Jardin botanique, Herbier, 69205 Lyon cedex 01, France - [email protected] Citation : Danet F., 2019. Alphabetical lists of the vascular plant families with their phylogenetic classification numbers. Colligo, 2(1) : 3- 10. https://perma.cc/2WFD-A2A7 KEY-WORDS Angiosperms family arrangement Summary: This paper provides, for herbarium cura- Gymnosperms Classification tors, the alphabetical lists of the recognized families Pteridophytes APG system in pteridophytes, gymnosperms and angiosperms Ferns PPG system with their phylogenetic classification numbers. Lycophytes phylogeny Herbarium MOTS-CLÉS Angiospermes rangement des familles Résumé : Cet article produit, pour les conservateurs Gymnospermes Classification d’herbier, les listes alphabétiques des familles recon- Ptéridophytes système APG nues pour les ptéridophytes, les gymnospermes et Fougères système PPG les angiospermes avec leurs numéros de classement Lycophytes phylogénie phylogénétique. Herbier Introduction These alphabetical lists have been established for the systems of A.-L de Jussieu, A.-P. de Can- The organization of herbarium collections con- dolle, Bentham & Hooker, etc. that are still used sists in arranging the specimens logically to in the management of historical herbaria find and reclassify them easily in the appro- whose original classification is voluntarily pre- priate storage units. In the vascular plant col- served. lections, commonly used methods are systema- Recent classification systems based on molecu- tic classification, alphabetical classification, or lar phylogenies have developed, and herbaria combinations of both. -

Perú: Cordillera Escalera-Loreto Perú: Cordillera Escalera-Loreto Escalera-Loreto Cordillera Perú: Instituciones Participantes/ Participating Institutions
.................................................................................................................................................................................................................................................................................................................................................................................................................................................................................................................... .............................................................................................................................................................................................................................................................................................................................................................................................no. 26 ....................................................................................................................... 26 Perú: Cordillera Escalera-Loreto Perú: Cordillera Escalera-Loreto Instituciones participantes/ Participating Institutions The Field Museum Nature and Culture International (NCI) Federación de Comunidades Nativas Chayahuita (FECONACHA) Organización Shawi del Yanayacu y Alto Paranapura (OSHAYAAP) Municipalidad Distrital de Balsapuerto Instituto de Investigaciones de la Amazonía Peruana (IIAP) Herbario Amazonense de la Universidad Nacional de la Amazonía Peruana (AMAZ) Museo de Historia Natural de la Universidad Nacional Mayor de San Marcos Centro -

The Occurrence of Red and Yellow Autumn Leaves Explained by Regional Differences in Insolation and Temperature
Review Tansley review The occurrence of red and yellow autumn leaves explained by regional differences in insolation and temperature Authors for correspondence: Susanne S. Renner1 and Constantin M. Zohner2 Susanne S. Renner 1 2 Tel: +49 89 17861 250 Systematic Botany and Mycology, University of Munich (LMU), Menzinger Str. 67, Munich 80638, Germany ; Institute of Email: [email protected] Integrative Biology, Department of Environmental Systems Science, ETH Zurich, Zurich 8092, Switzerland Constantin M. Zohner Email: [email protected] Received: 19 January 2019 Accepted: 24 April 2019 Contents Summary 1464 IV. The adaptive value of colour-changing leaves 1468 I. Introduction 1464 V. Outlook 1469 II. Phylogenetic and geographical occurrence of autumn colour Acknowledgements 1469 change 1465 References 1470 III. Physiological functions of autumnal leaf xanthophylls and anthocyanins 1466 Summary New Phytologist (2019) 224: 1464–1471 Red or yellow autumn leaves have long fascinated biologists, but their geographical doi: 10.1111/nph.15900 concentration in trees in Eastern North America (ENA) has defied evolutionary explanations. In this review, anthocyanins and xanthophylls are discussed in relation to their occurrence in Key words: adaptive explanation, different regions of the Northern Hemisphere, phylogenetic distribution and photoprotective anthocyanins, photo-oxidative damage, function during the breakdown of chlorophylls. Pigments in senescing leaves that intercept regional climates, solar irradiation, incident light and dissipate the absorbed energy extend the time available for nutrient resorption. xanthophylls. Experiments with Arabidopsis have revealed greatest anthocyanin photoprotective function at low temperatures and high light intensities, and high-resolution solar irradiation maps reveal that ENA and Asia receive higher irradiation than does Europe. -
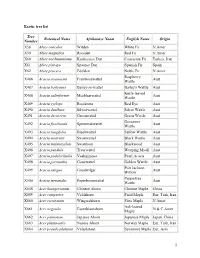
1 Exotic Tree List Tree Number Botanical Name Afrikaanse Naam
Exotic tree list Tree Botanical Name Afrikaanse Naam English Name Origin Number X58 Abies concolor Witden White Fir N.Amer X59 Abies magnifica Rooiden Red Fir N.Amer X60 Abies nordmanniana Kaukasiese Den Caucasian Fir Turkey, Iran X61 Abies pinsapo Spaanse Den Spanish Fir Spain X62 Abies procera Edelden Noble Fir N.Amer Raspberry X486 Acacia acuminata Frambosewattel Aust Wattle X487 Acacia baileyana Bailey-se-wattel Bailey's Wattle Aust Knife-leaved X488 Acacia cultriformis Mesblaarwattel Aust Wattle X489 Acacia cyclops Rooikrans Red Eye Aust X490 Acacia dealbata Silwerwattel Silver Wattle Aust X491 Acacia decurrens Groenwattel Green Wattle Aust Gossamer X492 Acacia floribunda Spinnerakwattel Aust Wattle X493 Acacia longifolia Bleekwattel Sallow Wattle Aust X494 Acacia mearnsii Swartwattel Black Wattle Aust X495 Acacia melanoxylon Swarthout Blackwood Aust X496 Acacia pendula Treurwattel Weeping Myall Aust X497 Acacia podalyriifolia Vaalmimosa Pearl Acacia Aust X498 Acacia pycnantha Gouewattel Golden Wattle Aust Port Jackson X499 Acacia saligna Goudwilger Aust Willow Peppertree X500 Acacia terminalis Peperboomwattel Aust Wattle X658 Acer buergerianum Chinese Ahorn Chinese Maple China X659 Acer campestre Veldahorn Field Maple Eur, Turk, Iran X660 Acer circinatum Wingerdahorn Vine Maple N Amer Ash-leaved X661 Acer negundo Essenblaarahorn N & C Amer Maple X662 Acer palmatum Japanse Ahorn Japanese Maple Japan, China X663 Acer platanoides Noorse Ahorn Norway Maple Eur, Turk, Iran X664 Acer pseudo-platanus Valsplataan Sycamore Maple Eur, Asia -

Evolutionary History of Floral Key Innovations in Angiosperms Elisabeth Reyes
Evolutionary history of floral key innovations in angiosperms Elisabeth Reyes To cite this version: Elisabeth Reyes. Evolutionary history of floral key innovations in angiosperms. Botanics. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS489. tel-01443353 HAL Id: tel-01443353 https://tel.archives-ouvertes.fr/tel-01443353 Submitted on 23 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS489 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N° 567 Sciences du Végétal : du Gène à l’Ecosystème Spécialité de Doctorat : Biologie Par Mme Elisabeth Reyes Evolutionary history of floral key innovations in angiosperms Thèse présentée et soutenue à Orsay, le 13 décembre 2016 : Composition du Jury : M. Ronse de Craene, Louis Directeur de recherche aux Jardins Rapporteur Botaniques Royaux d’Édimbourg M. Forest, Félix Directeur de recherche aux Jardins Rapporteur Botaniques Royaux de Kew Mme. Damerval, Catherine Directrice de recherche au Moulon Président du jury M. Lowry, Porter Curateur en chef aux Jardins Examinateur Botaniques du Missouri M. Haevermans, Thomas Maître de conférences au MNHN Examinateur Mme. Nadot, Sophie Professeur à l’Université Paris-Sud Directeur de thèse M. -

Phylogenomic Approach
Toward the ultimate phylogeny of Magnoliaceae: phylogenomic approach Sangtae Kim*1, Suhyeon Park1, and Jongsun Park2 1 Sungshin University, Korea 2 InfoBoss Co., Korea Mr. Carl Ferris Miller Founder of Chollipo Arboretum in Korea Chollipo Arboretum Famous for its magnolia collection 2020. Annual Meeting of Magnolia Society International Cholliop Arboretum in Korea. April 13th~22th, 2020 http://WWW.Chollipo.org Sungshin University, Seoul, Korea Dr. Hans Nooteboom Dr. Liu Yu-Hu Twenty-one years ago... in 1998 The 1st International Symposium on the Family Magnoliaceae, Gwangzhow Dr. Hiroshi Azuma Mr. Richard Figlar Dr. Hans Nooteboom Dr. Qing-wen Zeng Dr. Weibang Sun Handsome young boy Dr. Yong-kang Sima Dr. Yu-wu Law Presented ITS study on Magnoliaceae - never published Ten years ago... in 2009 Presented nine cp genome region study (9.2 kbp) on Magnoliaceae – published in 2013 2015 1st International Sympodium on Neotropical Magnoliaceae Gadalajara, 2019 3rd International Sympodium and Workshop on Neotropical Magnoliaceae Asterales Dipsacales Apiales Why magnolia study is Aquifoliales Campanulids (Euasterids II) Garryales Gentianales Laminales Solanales Lamiids important in botany? Ericales Asterids (Euasterids I) Cornales Sapindales Malvales Brassicales Malvids Fagales (Eurosids II) • As a member of early-diverging Cucurbitales Rosales Fabales Zygophyllales Celestrales Fabids (Eurosid I) angiosperms, reconstruction of the Oxalidales Malpighiales Vitales Geraniales Myrtales Rosids phylogeny of Magnoliaceae will Saxifragales Caryphyllales