Identification of 12 Cancer Types Through Genome Deep
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Genome Functional Annotation Across Species Using Deep Convolutional Neural Networks
bioRxiv preprint doi: https://doi.org/10.1101/330308; this version posted June 7, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome Functional Annotation across Species using Deep Convolutional Neural Networks Ghazaleh Khodabandelou1,*, Etienne Routhier1, and Julien Mozziconacci1,* 1Sorbonne Universite,´ CNRS, Laboratoire de Physique Theorique´ de la Matiere` Condensee,` LPTMC, 75005 Paris, France. *[email protected], [email protected] ABSTRACT Deep neural network application is today a skyrocketing field in many disciplinary domains. In genomics the development of deep neural networks is expected to revolutionize current practice. Several approaches relying on convolutional neural networks have been developed to associate short genomic sequences with a functional role such as promoters, enhancers or protein binding sites along genomes. These approaches rely on the generation of sequences batches with known annotations for learning purpose. While they show good performance to predict annotations from a test subset of these batches, they usually perform poorly when applied genome-wide. In this study, we address this issue and propose an optimal strategy to train convolutional neural networks for this specific application. We use as a case study transcription start sites and show that a model trained on one organism can be used to predict transcription start sites in a different specie. This cross-species application of convolutional neural networks trained with genomic sequence data provides a new technique to annotate any genome from previously existing annotations in related species. It also provides a way to determine whether the sequence patterns recognized by chromatin associated proteins in different species are conserved or not. -

Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal Through Mirna-1247-3P in Human Cervical Cancer Cells
Article Volume 11, Issue 1, 2021, 7943 - 7972 https://doi.org/10.33263/BRIAC111.79437972 Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal through miRNA-1247-3p in Human Cervical Cancer Cells Meng Luo 1 , Dian Fan 2 , Yinan Xiao 2 , Hao Zeng 2 , Dingyue Zhang 2 , Yunuo Zhao 2 , Xuelei Ma 1,* 1 Department of Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (L.M.); 2 West China Medical School, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (F.D.); [email protected] (X.Y.N.); [email protected] (Z.H.); [email protected] (Z.D.Y.); [email protected] (Z.Y.N.); * Correspondence: [email protected]; Scopus Author ID 55523405000 Received: 11.06.2020; Revised: 3.07.2020; Accepted: 5.07.2020; Published: 9.07.2020 Abstract: The prognosis of cervical cancer remains poor. Sulforaphane, an active ingredient from cruciferous plants, has been identified as a potential anticancer agent in various cancers. However, there is little information about its effect on cervical cancer. Here, we conducted a present study to uncover the effect and the potential mechanisms of sulforaphane on cervical cancer. HeLa cells were treated with sulforaphane, and cell proliferation and apoptosis were assessed by Cell Counting Kit-8, Western blot, flow cytometry, and immunofluorescence. Then, next-generation sequencing (NGS) and bioinformatics tools were used to analyze mRNA-seq, miRNA-seq, and potential pathways. Finally, qRT-PCR, Cell Counting Kit-8, flow cytometry, small RNAs analysis, and Western blot were performed to evaluate the biological function of miR1247-3p and MAPK pathway in HeLa cell lines. -

Table S4. RAE Analysis of Dedifferentiated Liposarcoma
Table S4. RAE analysis of dedifferentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes genes Amp 1 57809872 60413476 2603605 0.00026 34.6 10 DAB1,RPS26P15,OMA1,TACSTD2,MYSM1,JUN,FGGY,HOOK1,CYP2J2,C1orf87 Amp 1 158619146 158696968 77823 0.053 25 1 VANGL2 Amp 1 158883523 158922841 39319 0.081 23.1 2 SLAMF1,CD48 Amp 1 162042586 162118557 75972 0.072 25 0 [Nearest:NUF2] Amp 1 162272460 162767627 495168 0.017 26.9 0 [Nearest:PBX1] Amp 1 165486554 165532374 45821 0.057 25 1 POU2F1 Amp 1 167138282 167483267 344986 0.024 26.9 2 ATP1B1,NME7 Amp 1 167612872 167708844 95973 0.041 25 3 BLZF1,C1orf114,SLC19A2 Amp 1 167728199 167808161 79963 0.076 21.2 1 F5 Amp 1 168436370 169233893 797524 0.018 26.9 3 GORAB,PRRX1,C1orf129 Amp 1 169462231 170768440 1306210 1.3E-06 38.5 10 FMO1,FMO4,TOP1P1,BAT2D1,MYOC,VAMP4,METTL13,DNM3,C1orf105,PIGC Amp 1 171026247 171291427 265181 0.015 26.9 1 TNFSF18 Del 1 201860394 202299299 438906 0.0047 25 6 ATP2B4,SNORA77,LAX1,ZC3H11A,SNRPE,C1orf157 Del 1 210909187 212021116 1111930 0.017 19.2 8 BATF3,NSL1,TATDN3,C1orf227,FLVCR1,VASH2,ANGEL2,RPS6KC1 Del 1 215937857 216049214 111358 0.079 23.1 1 SPATA17 Del 1 218237257 218367476 130220 0.0063 26.9 3 EPRS,BPNT1,IARS2 Del 1 222100886 222727238 626353 5.2E-05 32.7 5 FBXO28,DEGS1,NVL,CNIH4,WDR26 Del 1 223166548 224519805 1353258 0.0063 26.9 15 DNAH14,LBR,ENAH,SRP9,EPHX1,TMEM63A,LEFTY1,PYCR2,LEFTY2,C1orf55,H3F3A,LOC440926 ,ACBD3,MIXL1,LIN9 Del 1 225283136 225374166 91031 0.054 23.1 1 CDC42BPA Del 1 227278990 229012661 1733672 0.091 21.2 13 RAB4A,SPHAR,C1orf96,ACTA1,NUP133,ABCB10,TAF5L,URB2,GALNT2,PGBD5,COG2,AGT,CAP -
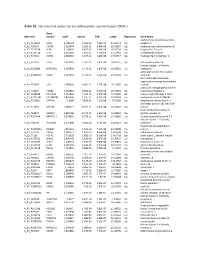
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Genome Functional Annotation Using Deep Convolutional Neural Network
bioRxiv preprint doi: https://doi.org/10.1101/330308; this version posted May 25, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome Functional Annotation using Deep Convolutional Neural Network Ghazaleh Khodabandelou1, Etienne Routhier1,2, Julien Mozziconacci1 1 Sorbonne Universit´e,CNRS, Laboratoire de Physique Th´eoriquede la Mati`ere Condens´ee,LPTMC, 75005 Paris, France. 2 ENS Cachan, Cachan, France * [email protected], [email protected] Abstract Deep neural network application is today a skyrocketing field in almost all disciplinary domains. In genomics, which deals with DNA sequences, the development of deep neural networks is expected to revolutionize current practice, from fundamental issues such as understanding the evolution of genomes to more specific applications such as the development of personalized medicine. Several approaches have been developed relying on convolution neural networks (CNN) to identify the functional role of sequences such as promoters, enhancers or protein binding sites along genomes. These approaches rely on the generation of sequences batches with known annotations for learning purpose. While they show good performance to predict annotations from a test subset of these batches, they usually work less well when applied genome-wide; i.e. for whole genome annotation. In this paper, we address this issue and propose an optimal strategy to train CNN for this specific application. We use as a case study gene Transcription Start Sites (TSS) and show that a model trained on one organism (e.g. human) can be used to predict TSS in a different specie (e.g. -

Mirnas Documented to Play a Role in Hematopoietic Cell Lineage. Our
Table S1: miRNAs documented to play a role in hematopoietic cell lineage. Our review of the literature summarizing miRNAs known to be involved in the development and proliferation of the hematopoietic lineage cells. miRNA Expression/function/target/regulator References miR-150 Elevated during developmental stages of B and T cell maturation. 16-19 Controls B cell differentiation, present in mature, resting T and B cells but decreased upon activation of naïve T or B cells. Plays a role in establishing lymphocyte identity. Very little is known about function in T cells. Regulators: Foxp3 Target: C-Myb miR-146a/b Upregulated in macropgahe inflammatory response. Differentially 17, 20 upregulated in murine Th1 subset but abolished in Th2 subset. Upregulated in response to TCRs stimulation, as well as by IL-1 and TNF. Highly expressed in murine T-regs and could play a role in establishing lymphocyte identity. Modulates activation induced cell death in activated T cells. Negative regulator of TLR and cytokine signaling pathway. Endotoxin tolerance. Antiviral role. Targets: IRAK1, IRAK2, TRAF6, FAF1 miR-16-1 Promote apoptosis by targeting Bcl2 expression, act as tumor 22 cluster suppressor RNAs. May block differentiation of later stage hematopoietic progenitor cells to mature cells. Downregulated in CLL. Target: BCL2. miR-155 Regulator of T and B cell maturation and innate immune response. 23-29 Expressed in primary mediastinal B-cell lymphoma, T and B cells, macrophages and DCs. Upregulated during B cell activation. Involved in T cell differentiation and indicated as a positive regulator of cytokine production. Activated by stimulating TLR3 and INFab receptors in bone derived macrophages (regulation of antimicrobial defense). -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -

Downloaded from (CCLE) Web Site (
bioRxiv preprint doi: https://doi.org/10.1101/852939; this version posted January 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Patel, A et al. 1 Integrative genomic and epigenomic analyses identify a distinct role of c-Myc and L-Myc for lineage determination in small cell lung cancer Ayushi S. Patel1,2, Seungyeul Yoo3,4, Ranran Kong1,2,5, Takashi Sato1,2,6, Maya Fridrikh1,2, Abhilasha Sinha1,2, German Nudelman7, Charles A. Powell1,2, Mary Beth Beasley8, Jun Zhu2,3,4, Hideo Watanabe1,2,3,9* 1Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA 2Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA 3Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA 4Sema4, a Mount Sinai venture, Stamford, CT 06902, USA. 5Department of Thoracic Surgery, The Second Affiliated Hospital of Medical School, Xi'an Jiaotong University, Xi'an, Shaanxi, 710004, China 6Division of Pulmonary Medicine, Department of Medicine, Keio University School of Medicine, 35 Shinanomachi, Shinjuku, Tokyo 160-8582, Japan 7 Department of Neurology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA 8Department of Pathology and Laboratory Medicine, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA 9Lead Contact *Correspondence: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/852939; this version posted January 23, 2020. -

Supplemental Table 3 Two-Class Paired Significance Analysis of Microarrays Comparing Gene Expression Between Paired
Supplemental Table 3 Two‐class paired Significance Analysis of Microarrays comparing gene expression between paired pre‐ and post‐transplant kidneys biopsies (N=8). Entrez Fold q‐value Probe Set ID Gene Symbol Unigene Name Score Gene ID Difference (%) Probe sets higher expressed in post‐transplant biopsies in paired analysis (N=1871) 218870_at 55843 ARHGAP15 Rho GTPase activating protein 15 7,01 3,99 0,00 205304_s_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 6,30 4,50 0,00 1563649_at ‐‐ ‐‐ ‐‐ 6,24 3,51 0,00 1567913_at 541466 CT45‐1 cancer/testis antigen CT45‐1 5,90 4,21 0,00 203932_at 3109 HLA‐DMB major histocompatibility complex, class II, DM beta 5,83 3,20 0,00 204606_at 6366 CCL21 chemokine (C‐C motif) ligand 21 5,82 10,42 0,00 205898_at 1524 CX3CR1 chemokine (C‐X3‐C motif) receptor 1 5,74 8,50 0,00 205303_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 5,68 6,87 0,00 226841_at 219972 MPEG1 macrophage expressed gene 1 5,59 3,76 0,00 203923_s_at 1536 CYBB cytochrome b‐245, beta polypeptide (chronic granulomatous disease) 5,58 4,70 0,00 210135_s_at 6474 SHOX2 short stature homeobox 2 5,53 5,58 0,00 1562642_at ‐‐ ‐‐ ‐‐ 5,42 5,03 0,00 242605_at 1634 DCN decorin 5,23 3,92 0,00 228750_at ‐‐ ‐‐ ‐‐ 5,21 7,22 0,00 collagen, type III, alpha 1 (Ehlers‐Danlos syndrome type IV, autosomal 201852_x_at 1281 COL3A1 dominant) 5,10 8,46 0,00 3493///3 IGHA1///IGHA immunoglobulin heavy constant alpha 1///immunoglobulin heavy 217022_s_at 494 2 constant alpha 2 (A2m marker) 5,07 9,53 0,00 1 202311_s_at -

Identification and Interaction Analysis of Molecular Markers in Pancreatic
medRxiv preprint doi: https://doi.org/10.1101/2020.12.20.20248601; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification and Interaction Analysis of Molecular Markers in Pancreatic Ductal Adenocarcinoma by Integrated Bioinformatics Analysis and Molecular Docking Experiments Basavaraj Vastrad1 , Chanabasayya Vastrad *2, Anandkumar Tengli3 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics,Chanabasava Nilaya, Bharthinagar, Dharwad, Karanataka 580001, India. 3. Department of Pharmaceutical Chemistry, JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education & Research, Mysuru- 570015, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.20.20248601; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract The current investigation aimed to mine therapeutic molecular targets that play an key part in the advancement of pancreatic ductal adenocarcinoma (PDAC). The expression profiling by high throughput sequencing dataset profile GSE133684 dataset was downloaded from the Gene Expression Omnibus (GEO) database.