High-Resolution Characterization of the Leukemic Cutaneous T-Cell Lymphoma Genome William Lin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Database Tool the Systematic Annotation of the Three Main GPCR
Database, Vol. 2010, Article ID baq018, doi:10.1093/database/baq018 ............................................................................................................................................................................................................................................................................................. Database tool The systematic annotation of the three main Downloaded from https://academic.oup.com/database/article-abstract/doi/10.1093/database/baq018/406672 by guest on 15 January 2019 GPCR families in Reactome Bijay Jassal1, Steven Jupe1, Michael Caudy2, Ewan Birney1, Lincoln Stein2, Henning Hermjakob1 and Peter D’Eustachio3,* 1European Bioinformatics Institute, Hinxton, Cambridge, CB10 1SD, UK, 2Ontario Institute for Cancer Research, Toronto, ON M5G 0A3, Canada and 3New York University School of Medicine, New York, NY 10016, USA *Corresponding author: Tel: +212 263 5779; Fax: +212 263 8166; Email: [email protected] Submitted 14 April 2010; Revised 14 June 2010; Accepted 13 July 2010 ............................................................................................................................................................................................................................................................................................. Reactome is an open-source, freely available database of human biological pathways and processes. A major goal of our work is to provide an integrated view of cellular signalling processes that spans from ligand–receptor -

Structure of the Tripartite Motif of KAP1/TRIM28 Identifies Molecular Interfaces Required for Transcriptional Silencing of Retrotransposons
bioRxiv preprint doi: https://doi.org/10.1101/505677; this version posted December 25, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Structure of the tripartite motif of KAP1/TRIM28 identifies molecular interfaces required for transcriptional silencing of retrotransposons Guido A. Stoll1, Shun-ichiro Oda1, Zheng-Shan Chong1, Minmin Yu2, Stephen H. McLaughlin2 and Yorgo Modis1,* 1 Molecular Immunity Unit, Department of Medicine, University of Cambridge, MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus, Cambridge, CB2 0QH, UK 2 MRC Laboratory oF Molecular Biology, Cambridge Biomedical Campus, Cambridge, CB2 0QH, UK * Correspondence: [email protected] (Y.M.) Abstract Transcription oF transposable elements is tightly regulated to prevent damage to the genome. The family of KRAB domain-containing zinc Finger proteins (KRAB-ZFPs) and KRAB-associated protein 1 (KAP1/TRIM28) play a key role in regulating retrotransposons. KRAB-ZFPs recognize speciFic retrotransposon sequences and recruit KAP1, which controls the assembly of an epigenetic silencing complex including histone H3K9 methyltransferase SETDB1. The chromatin remodeling activities of this complex repress transcription of the targeted transposable element and any adjacent genes. Here, we use biophysical and structural approaches to show that the tripartite motif (TRIM) of KAP1 Forms antiparallel dimers, which Further assemble into tetramers and higher-order oligomers in a concentration-dependent manner. Structure-based mutations in the B-box 1 domain prevented higher-order oligomerization without a signiFicant loss oF retrotransposon silencing activity in a cell-based assay, indicating that, in contrast to other TRIM family members, selF-assembly is not essential For the function of KAP1. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Human Artificial Chromosome (Hac) Vector
Europäisches Patentamt *EP001559782A1* (19) European Patent Office Office européen des brevets (11) EP 1 559 782 A1 (12) EUROPEAN PATENT APPLICATION published in accordance with Art. 158(3) EPC (43) Date of publication: (51) Int Cl.7: C12N 15/09, C12N 1/15, 03.08.2005 Bulletin 2005/31 C12N 1/19, C12N 1/21, C12N 5/10, C12P 21/02 (21) Application number: 03751334.8 (86) International application number: (22) Date of filing: 03.10.2003 PCT/JP2003/012734 (87) International publication number: WO 2004/031385 (15.04.2004 Gazette 2004/16) (84) Designated Contracting States: • KATOH, Motonobu, Tottori University AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Yonago-shi, Tottori 683-8503 (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • TOMIZUKA, Kazuma, Designated Extension States: Kirin Beer Kabushiki Kaisha AL LT LV MK Takashi-shi, Gunma 370-1295 (JP) • KUROIWA, Yoshimi, (30) Priority: 04.10.2002 JP 2002292853 Kirin Beer Kabushiki Kaisha Takasaki-shi, Gunma 370-1295 (JP) (71) Applicant: KIRIN BEER KABUSHIKI KAISHA • KAKEDA, Minoru, Kirin Beer Kabushiki Kaisha Tokyo 104-8288 (JP) Takasaki-shi, Gunma 370-1295 (JP) (72) Inventors: (74) Representative: HOFFMANN - EITLE • OSHIMURA, Mitsuo, Tottori University Patent- und Rechtsanwälte Yonago-shi, Tottori 683-8503 (JP) Arabellastrasse 4 81925 München (DE) (54) HUMAN ARTIFICIAL CHROMOSOME (HAC) VECTOR (57) The present invention relates to a human arti- ing a cell which expresses foreign DNA. Furthermore, ficial chromosome (HAC) vector and a method for pro- the present invention relates to a method for producing ducing the same. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Screening of Differentially Expressed Microsporidia Genes From
insects Article Screening of Differentially Expressed Microsporidia Genes from Nosema ceranae Infected Honey Bees by Suppression Subtractive Hybridization 1, 1 2 1, 3, , Zih-Ting Chang y, Chong-Yu Ko , Ming-Ren Yen , Yue-Wen Chen * and Yu-Shin Nai * y 1 Department of Biotechnology and Animal Science, National Ilan University, No. 1, Sec. 1, Shen Nung Road, Ilan 26047, Taiwan; [email protected] (Z.-T.C.); [email protected] (C.-Y.K.) 2 Genomics Research Center, Academia Sinica, No. 128, Academia Road, Sec. 2, Nankang District, Taipei 115, Taiwan; [email protected] 3 Department of Entomology, National Chung Hsing University, No. 145, Xingda Road, Taichung 402, Taiwan * Correspondence: [email protected] (Y.-W.C.); [email protected] (Y.-S.N.) These authors contributed equally to this work. y Received: 21 February 2020; Accepted: 18 March 2020; Published: 22 March 2020 Abstract: The microsporidium Nosema ceranae is a high prevalent parasite of the European honey bee (Apis mellifera). This parasite is spreading across the world into its novel host. The developmental process, and some mechanisms of N. ceranae-infected honey bees, has been studied thoroughly; however, few studies have been carried out in the mechanism of gene expression in N. ceranae during the infection process. We therefore performed the suppressive subtractive hybridization (SSH) approach to investigate the candidate genes of N. ceranae during its infection process. All 96 clones of infected (forward) and non-infected (reverse) library were dipped onto the membrane for hybridization. A total of 112 differentially expressed sequence tags (ESTs) had been sequenced. -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Greg's Awesome Thesis
Analysis of alignment error and sitewise constraint in mammalian comparative genomics Gregory Jordan European Bioinformatics Institute University of Cambridge A dissertation submitted for the degree of Doctor of Philosophy November 30, 2011 To my parents, who kept us thinking and playing This dissertation is the result of my own work and includes nothing which is the out- come of work done in collaboration except where specifically indicated in the text and acknowledgements. This dissertation is not substantially the same as any I have submitted for a degree, diploma or other qualification at any other university, and no part has already been, or is currently being submitted for any degree, diploma or other qualification. This dissertation does not exceed the specified length limit of 60,000 words as defined by the Biology Degree Committee. November 30, 2011 Gregory Jordan ii Analysis of alignment error and sitewise constraint in mammalian comparative genomics Summary Gregory Jordan November 30, 2011 Darwin College Insight into the evolution of protein-coding genes can be gained from the use of phylogenetic codon models. Recently sequenced mammalian genomes and powerful analysis methods developed over the past decade provide the potential to globally measure the impact of natural selection on pro- tein sequences at a fine scale. The detection of positive selection in particular is of great interest, with relevance to the study of host-parasite conflicts, immune system evolution and adaptive dif- ferences between species. This thesis examines the performance of methods for detecting positive selection first with a series of simulation experiments, and then with two empirical studies in mammals and primates. -

Microarray-Based Analysis of Genes, Transcription Factors, and Epigenetic Modifications in Lung Cancer Exposed to Nitric Oxide
CANCER GENOMICS & PROTEOMICS 17 : 401-415 (2020) doi:10.21873/cgp.20199 Microarray-based Analysis of Genes, Transcription Factors, and Epigenetic Modifications in Lung Cancer Exposed to Nitric Oxide ARNATCHAI MAIUTHED 1, ORNJIRA PRAKHONGCHEEP 2,3 and PITHI CHANVORACHOTE 2,3 1Department of Pharmacology, Faculty of Pharmacy, Mahidol University, Bangkok, Thailand; 2Cell-based Drug and Health Product Development Research Unit, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand; 3Department of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand Abstract. Background/Aim: Nitric oxide (NO) is recognized NO exposure of lung cancer cells resulted in a change in as an important biological mediator that exerts several transcription factors (TFs) and epigenetic modifications human physiological functions. As its nature is an aqueous (histone modification and miRNA). Interestingly, NO soluble gas that can diffuse through cells and tissues, NO treatment was shown to potentiate cancer stem cell-related can affect cell signaling, the phenotype of cancer and modify genes and transcription factors Oct4, Klf4, and Myc. surrounding cells. The variety of effects of NO on cancer cell Conclusion: Through this comprehensive approach, the biology has convinced researchers to determine the defined present study illustrated the scheme of how NO affects mechanisms of these effects and how to control this mediator molecular events in lung cancer cells. for a better understanding as well as for therapeutic gain. Materials and Methods: We used bioinformatics and Lung cancer is one of the leading causes of cancer-related pharmacological experiments to elucidate the potential deaths worldwide (1, 2). Since the aggressive phenotypes regulation and underlying mechanisms of NO in non-small and treatment failures have been frequently observed in lung a lung cancer cell model. -

Supplemental Figure S1 Differentially Methylated Regions (Dmrs
Supplemental Figure S1 '$$#0#,2'**7+#2&7*2#"0#%'-,11 #25##,"'1#1#122#1 '!2-0'*"#.'!2'-,-$122,1'2'-,$0-+2- !"Q !"2-$%," $ 31',% 25-$-*" !&,%# ," ' 0RTRW 1 !32V-$$ !0'2#0'T - #.0#1#,22'-, -$ "'$$#0#,2'**7+#2&7*2#"%#,#11',.0#,2#1,"2&#'0 #&4'-022,1'2'-, #25##,"'$$#0#,2"'1#1#122#1T-*!)00-51',"'!2#&7.#0+#2&7*2#"%#,#1Q%0700-51 &7.-+#2&7*2#"%#,#1Q31',%25-$-*"!&,%#,"'0RTRW1!32V-$$!0'2#0'T-%#,#1 +#22&# -4#!0'2#0'22,1'2'-,$0-+$%2-$Q5#2�#$-0#*1-',!*3"#" %#,#15'2&V4*3#0RTRWT$$#!2#"%#,10#&'%&*'%&2#" 712#0'1)1#T Supplemental Figure S2 Validation of results from the HELP assay using Epityper MassarrayT #13*21 $0-+ 2&# 1$ 117 5#0# !-00#*2#" 5'2& /3,2'22'4# +#2&7*2'-, ,*78#" 7 '13*$'2#11007$-04V-,"6U-%#,#.0-+-2#00#%'-,1T11007 51.#0$-0+#"31',%**4'* *#1+.*#1T S Supplemental Fig. S1 A unique hypermethylated genes (methylation sites) 454 (481) 5693 (6747) 120 (122) NLMGUS NEWMM REL 2963 (3207) 1338 (1560) 5 (5) unique hypomethylated genes (methylation sites) B NEWMM 0 (0) MGUS 454 (481) 0 (0) NEWMM REL NL 3* (2) 2472 (3066) NEWMM 2963 REL (3207) 2* (2) MGUS 0 (0) REL 2 (2) NEWMM 0 (0) REL Supplemental Fig. S2 A B ARID4B DNMT3A Methylation by MassArray Methylation by MassArray 0 0.2 0.4 0.6 0.8 1 1.2 0.5 0.6 0.7 0.8 0.9 1 2 0 NL PC MGUS 1.5 -0.5 NEW MM 1 REL MM -1 0.5 -1.5 0 -2 -0.5 -1 -2.5 -1.5 -3 Methylation by HELP Assay Methylation by HELP Methylation by HELP Assay Methylation by HELP -2 -3.5 -2.5 -4 Supplemental tables "3..*#+#,2*6 *#"SS 9*','!*!&0!2#0'12'!1-$.2'#,21+.*#1 DZ_STAGE Age Gender Ethnicity MM isotype PCLI Cytogenetics -

Identification of Candidate Biomarkers and Pathways Associated with Type 1 Diabetes Mellitus Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 1 diabetes mellitus (T1DM) is a metabolic disorder for which the underlying molecular mechanisms remain largely unclear. This investigation aimed to elucidate essential candidate genes and pathways in T1DM by integrated bioinformatics analysis. In this study, differentially expressed genes (DEGs) were analyzed using DESeq2 of R package from GSE162689 of the Gene Expression Omnibus (GEO). Gene ontology (GO) enrichment analysis, REACTOME pathway enrichment analysis, and construction and analysis of protein-protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network, and validation of hub genes were then performed. A total of 952 DEGs (477 up regulated and 475 down regulated genes) were identified in T1DM. GO and REACTOME enrichment result results showed that DEGs mainly enriched in multicellular organism development, detection of stimulus, diseases of signal transduction by growth factor receptors and second messengers, and olfactory signaling pathway. -
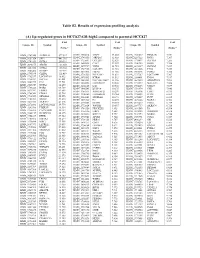
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688