Genome Functional Annotation Across Species Using Deep Convolutional Neural Networks
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Genome-Wide DNA Methylation Analysis on C-Reactive Protein Among Ghanaians Suggests Molecular Links to the Emerging Risk of Cardiovascular Diseases ✉ Felix P
www.nature.com/npjgenmed ARTICLE OPEN Genome-wide DNA methylation analysis on C-reactive protein among Ghanaians suggests molecular links to the emerging risk of cardiovascular diseases ✉ Felix P. Chilunga 1 , Peter Henneman2, Andrea Venema2, Karlijn A. C. Meeks 3, Ana Requena-Méndez4,5, Erik Beune1, Frank P. Mockenhaupt6, Liam Smeeth7, Silver Bahendeka8, Ina Danquah9, Kerstin Klipstein-Grobusch10,11, Adebowale Adeyemo 3, Marcel M.A.M Mannens2 and Charles Agyemang1 Molecular mechanisms at the intersection of inflammation and cardiovascular diseases (CVD) among Africans are still unknown. We performed an epigenome-wide association study to identify loci associated with serum C-reactive protein (marker of inflammation) among Ghanaians and further assessed whether differentially methylated positions (DMPs) were linked to CVD in previous reports, or to estimated CVD risk in the same population. We used the Illumina Infinium® HumanMethylation450 BeadChip to obtain DNAm profiles of blood samples in 589 Ghanaians from the RODAM study (without acute infections, not taking anti-inflammatory medications, CRP levels < 40 mg/L). We then used linear models to identify DMPs associated with CRP concentrations. Post-hoc, we evaluated associations of identified DMPs with elevated CVD risk estimated via ASCVD risk score. We also performed subset analyses at CRP levels ≤10 mg/L and replication analyses on candidate probes. Finally, we assessed for biological relevance of our findings in public databases. We subsequently identified 14 novel DMPs associated with CRP. In post-hoc evaluations, we found 1234567890():,; that DMPs in PC, BTG4 and PADI1 showed trends of associations with estimated CVD risk, we identified a separate DMP in MORC2 that was associated with CRP levels ≤10 mg/L, and we successfully replicated 65 (24%) of previously reported DMPs. -

The HSP70 Chaperone Machinery: J Proteins As Drivers of Functional Specificity
REVIEWS The HSP70 chaperone machinery: J proteins as drivers of functional specificity Harm H. Kampinga* and Elizabeth A. Craig‡ Abstract | Heat shock 70 kDa proteins (HSP70s) are ubiquitous molecular chaperones that function in a myriad of biological processes, modulating polypeptide folding, degradation and translocation across membranes, and protein–protein interactions. This multitude of roles is not easily reconciled with the universality of the activity of HSP70s in ATP-dependent client protein-binding and release cycles. Much of the functional diversity of the HSP70s is driven by a diverse class of cofactors: J proteins. Often, multiple J proteins function with a single HSP70. Some target HSP70 activity to clients at precise locations in cells and others bind client proteins directly, thereby delivering specific clients to HSP70 and directly determining their fate. In their native cellular environment, polypeptides are participates in such diverse cellular functions. Their constantly at risk of attaining conformations that pre- functional diversity is remarkable considering that vent them from functioning properly and/or cause them within and across species, HSP70s have high sequence to aggregate into large, potentially cytotoxic complexes. identity. They share a single biochemical activity: an Molecular chaperones guide the conformation of proteins ATP-dependent client-binding and release cycle com- throughout their lifetime, preventing their aggregation bined with client protein recognition, which is typi- by protecting interactive surfaces against non-productive cally rather promiscuous. This apparent conundrum interactions. Through such inter actions, molecular chap- is resolved by the fact that HSP70s do not work alone, erones aid in the folding of nascent proteins as they are but rather as ‘HSP70 machines’, collaborating with synthesized by ribosomes, drive protein transport across and being regulated by several cofactors. -

Prognostic and Functional Significant of Heat Shock Proteins (Hsps)
biology Article Prognostic and Functional Significant of Heat Shock Proteins (HSPs) in Breast Cancer Unveiled by Multi-Omics Approaches Miriam Buttacavoli 1,†, Gianluca Di Cara 1,†, Cesare D’Amico 1, Fabiana Geraci 1 , Ida Pucci-Minafra 2, Salvatore Feo 1 and Patrizia Cancemi 1,2,* 1 Department of Biological Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, 90128 Palermo, Italy; [email protected] (M.B.); [email protected] (G.D.C.); [email protected] (C.D.); [email protected] (F.G.); [email protected] (S.F.) 2 Experimental Center of Onco Biology (COBS), 90145 Palermo, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-091-2389-7330 † These authors contributed equally to this work. Simple Summary: In this study, we investigated the expression pattern and prognostic significance of the heat shock proteins (HSPs) family members in breast cancer (BC) by using several bioinfor- matics tools and proteomics investigations. Our results demonstrated that, collectively, HSPs were deregulated in BC, acting as both oncogene and onco-suppressor genes. In particular, two different HSP-clusters were significantly associated with a poor or good prognosis. Interestingly, the HSPs deregulation impacted gene expression and miRNAs regulation that, in turn, affected important bio- logical pathways involved in cell cycle, DNA replication, and receptors-mediated signaling. Finally, the proteomic identification of several HSPs members and isoforms revealed much more complexity Citation: Buttacavoli, M.; Di Cara, of HSPs roles in BC and showed that their expression is quite variable among patients. In conclusion, G.; D’Amico, C.; Geraci, F.; we elaborated two panels of HSPs that could be further explored as potential biomarkers for BC Pucci-Minafra, I.; Feo, S.; Cancemi, P. -

Morphology, Behavior, and the Sonic Hedgehog Pathway in Mouse Models of Down Syndrome
MORPHOLOGY, BEHAVIOR, AND THE SONIC HEDGEHOG PATHWAY IN MOUSE MODELS OF DOWN SYNDROME by Tara Dutka A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2014 © 2014 Tara Dutka All Rights Reserved Abstract Down Syndrome (DS) is caused by a triplication of human chromosome 21 (Hsa21). Ts65Dn, a mouse model of DS, contains a freely segregating extra chromosome consisting of the distal portion of mouse chromosome 16 (Mmu16), a region orthologous to part of Hsa21, and a non-Hsa21 orthologous region of mouse chromosome 17. All individuals with DS display some level of craniofacial dysmorphology, brain structural and functional changes, and cognitive impairment. Ts65Dn recapitulates these features of DS and aspects of each of these traits have been linked in Ts65Dn to a reduced response to Sonic Hedgehog (SHH) in trisomic cells. Dp(16)1Yey is a new mouse model of DS which has a direct duplication of the entire Hsa21 orthologous region of Mmu16. Dp(16)1Yey’s creators found similar behavioral deficits to those seen in Ts65Dn. We performed a quantitative investigation of the skull and brain of Dp(16)1Yey as compared to Ts65Dn and found that DS-like changes to brain and craniofacial morphology were similar in both models. Our results validate examination of the genetic basis for these phenotypes in Dp(16)1Yey mice and the genetic links for these phenotypes previously found in Ts65Dn , i.e., reduced response to SHH. Further, we hypothesized that if all trisomic cells show a reduced response to SHH, then up-regulation of the SHH pathway might ameliorate multiple phenotypes. -

Supplementary Appendix Table of Contents
Supplementary Appendix This appendix has been provided by the authors to give readers additional information about their work. Table of Contents Supplementary Note ........................................................................................................................... 1 Author contributions ............................................................................................................................... 1 Additional authors from study groups .............................................................................................. 2 Task Force COVID-19 Humanitas ......................................................................................................................................... 2 TASK FORCE COVID-19 HUMANITAS GAVAZZENI & CASTELLI ............................................................................. 3 COVICAT Study Group ............................................................................................................................................................... 4 Pa COVID-19 Study Group ....................................................................................................................................................... 5 Covid-19 Aachen Study (COVAS) .......................................................................................................................................... 5 Norwegian SARS-CoV-2 Study group ................................................................................................................................. -

Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal Through Mirna-1247-3P in Human Cervical Cancer Cells
Article Volume 11, Issue 1, 2021, 7943 - 7972 https://doi.org/10.33263/BRIAC111.79437972 Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal through miRNA-1247-3p in Human Cervical Cancer Cells Meng Luo 1 , Dian Fan 2 , Yinan Xiao 2 , Hao Zeng 2 , Dingyue Zhang 2 , Yunuo Zhao 2 , Xuelei Ma 1,* 1 Department of Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (L.M.); 2 West China Medical School, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (F.D.); [email protected] (X.Y.N.); [email protected] (Z.H.); [email protected] (Z.D.Y.); [email protected] (Z.Y.N.); * Correspondence: [email protected]; Scopus Author ID 55523405000 Received: 11.06.2020; Revised: 3.07.2020; Accepted: 5.07.2020; Published: 9.07.2020 Abstract: The prognosis of cervical cancer remains poor. Sulforaphane, an active ingredient from cruciferous plants, has been identified as a potential anticancer agent in various cancers. However, there is little information about its effect on cervical cancer. Here, we conducted a present study to uncover the effect and the potential mechanisms of sulforaphane on cervical cancer. HeLa cells were treated with sulforaphane, and cell proliferation and apoptosis were assessed by Cell Counting Kit-8, Western blot, flow cytometry, and immunofluorescence. Then, next-generation sequencing (NGS) and bioinformatics tools were used to analyze mRNA-seq, miRNA-seq, and potential pathways. Finally, qRT-PCR, Cell Counting Kit-8, flow cytometry, small RNAs analysis, and Western blot were performed to evaluate the biological function of miR1247-3p and MAPK pathway in HeLa cell lines. -

Table S4. RAE Analysis of Dedifferentiated Liposarcoma
Table S4. RAE analysis of dedifferentiated liposarcoma Model Chromosome Region start Region end Size q value freqX0* # genes genes Amp 1 57809872 60413476 2603605 0.00026 34.6 10 DAB1,RPS26P15,OMA1,TACSTD2,MYSM1,JUN,FGGY,HOOK1,CYP2J2,C1orf87 Amp 1 158619146 158696968 77823 0.053 25 1 VANGL2 Amp 1 158883523 158922841 39319 0.081 23.1 2 SLAMF1,CD48 Amp 1 162042586 162118557 75972 0.072 25 0 [Nearest:NUF2] Amp 1 162272460 162767627 495168 0.017 26.9 0 [Nearest:PBX1] Amp 1 165486554 165532374 45821 0.057 25 1 POU2F1 Amp 1 167138282 167483267 344986 0.024 26.9 2 ATP1B1,NME7 Amp 1 167612872 167708844 95973 0.041 25 3 BLZF1,C1orf114,SLC19A2 Amp 1 167728199 167808161 79963 0.076 21.2 1 F5 Amp 1 168436370 169233893 797524 0.018 26.9 3 GORAB,PRRX1,C1orf129 Amp 1 169462231 170768440 1306210 1.3E-06 38.5 10 FMO1,FMO4,TOP1P1,BAT2D1,MYOC,VAMP4,METTL13,DNM3,C1orf105,PIGC Amp 1 171026247 171291427 265181 0.015 26.9 1 TNFSF18 Del 1 201860394 202299299 438906 0.0047 25 6 ATP2B4,SNORA77,LAX1,ZC3H11A,SNRPE,C1orf157 Del 1 210909187 212021116 1111930 0.017 19.2 8 BATF3,NSL1,TATDN3,C1orf227,FLVCR1,VASH2,ANGEL2,RPS6KC1 Del 1 215937857 216049214 111358 0.079 23.1 1 SPATA17 Del 1 218237257 218367476 130220 0.0063 26.9 3 EPRS,BPNT1,IARS2 Del 1 222100886 222727238 626353 5.2E-05 32.7 5 FBXO28,DEGS1,NVL,CNIH4,WDR26 Del 1 223166548 224519805 1353258 0.0063 26.9 15 DNAH14,LBR,ENAH,SRP9,EPHX1,TMEM63A,LEFTY1,PYCR2,LEFTY2,C1orf55,H3F3A,LOC440926 ,ACBD3,MIXL1,LIN9 Del 1 225283136 225374166 91031 0.054 23.1 1 CDC42BPA Del 1 227278990 229012661 1733672 0.091 21.2 13 RAB4A,SPHAR,C1orf96,ACTA1,NUP133,ABCB10,TAF5L,URB2,GALNT2,PGBD5,COG2,AGT,CAP -

Actionable Druggable Genome-Wide Mendelian Randomization Identifies Repurposing Opportunities for COVID-19
ARTICLES https://doi.org/10.1038/s41591-021-01310-z Actionable druggable genome-wide Mendelian randomization identifies repurposing opportunities for COVID-19 Liam Gaziano1,2, Claudia Giambartolomei 3,4, Alexandre C. Pereira5,6, Anna Gaulton 7, Daniel C. Posner 1, Sonja A. Swanson8, Yuk-Lam Ho1, Sudha K. Iyengar9,10, Nicole M. Kosik 1, Marijana Vujkovic 11,12, David R. Gagnon 1,13, A. Patrícia Bento 7, Inigo Barrio-Hernandez14, Lars Rönnblom 15, Niklas Hagberg 15, Christian Lundtoft 15, Claudia Langenberg 16,17, Maik Pietzner 17, Dennis Valentine18,19, Stefano Gustincich 3, Gian Gaetano Tartaglia 3, Elias Allara 2, Praveen Surendran2,20,21,22, Stephen Burgess 2,23, Jing Hua Zhao2, James E. Peters 21,24, Bram P. Prins 2,21, Emanuele Di Angelantonio2,20,21,25,26, Poornima Devineni1, Yunling Shi1, Kristine E. Lynch27,28, Scott L. DuVall27,28, Helene Garcon1, Lauren O. Thomann1, Jin J. Zhou29,30, Bryan R. Gorman1, Jennifer E. Huffman 31, Christopher J. O’Donnell 32,33, Philip S. Tsao34,35, Jean C. Beckham36,37, Saiju Pyarajan1, Sumitra Muralidhar38, Grant D. Huang38, Rachel Ramoni38, Pedro Beltrao 14, John Danesh2,20,21,25,26, Adriana M. Hung39,40, Kyong-Mi Chang 12,41, Yan V. Sun 42,43, Jacob Joseph1,44, Andrew R. Leach7, Todd L. Edwards45,46, Kelly Cho1,47, J. Michael Gaziano1,47, Adam S. Butterworth 2,20,21,25,26 ✉ , Juan P. Casas1,47 ✉ and VA Million Veteran Program COVID-19 Science Initiative* Drug repurposing provides a rapid approach to meet the urgent need for therapeutics to address COVID-19. To identify thera- peutic targets relevant to COVID-19, we conducted Mendelian randomization analyses, deriving genetic instruments based on transcriptomic and proteomic data for 1,263 actionable proteins that are targeted by approved drugs or in clinical phase of drug development. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -
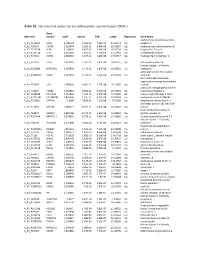
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454