View PDF Version
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hormonal Regulation During Initial Berry Development in Grapevine
1 Année 2013 Thèse N° 2064 THESE pour le DOCTORAT DE L’UNIVERSITE BORDEAUX 2 Ecole Doctorale des Sciences de la Vie et de la Santé de l’Université Bordeaux-Segalen Spécialité: Biologie Végétale Hormonal regulation during initial berry development in grapevine par María Francisca Godoy Santin Née le 20 Février 1985 à Santiago, Chili Soutenue publiquement le 15 Novembre, à Pontificia Universidad Católica de Chile, Santiago, Chili Membres du Jury : Pr. Alexis KALERGIS, Universidad Católica de Chile Pr. Michael HANDFORD, University of Chile Rapporteur Pr. Claudia STANGE, University of Chile Rapportrice Pr. Loreto HOLUIGUE, Universidad Católica de Chile Examinatrice Pr. Serge DELROT, Université Victor Ségalen Bordeaux2 Examinateur Pr. Patricio ARCE-JONHSON, Universidad Católica de Chile Co-Directeur de thèse Dr. Virginie LAUVERGEAT, Université Bordeaux1 Co-Directrice de thèse 2 Acknowledgements I would like to thank my advisors Dr Patricio Arce and Dr Virginie Lauvergeat for their guidance, patience and help at every step of the way. I also want to specially acknowledge to all my labmates for their ideas, support, laughs and much more: Jenn, Xime, Amparo, Mindy, Anita, Felipe, Tomás, Susan, Mónica, Jessy, Claudia, Dani H, and everyone else who made the working place the best one ever. Special thanks to Consuelo, who guided me from the first day, and to Anibal, who has been an amazing help during these lasts years. I want to thank Nathalie Kühn for all her ideas, discussion, company and help during these experiments, where she played a fundamental part. Also, I am very grateful to my other lab across the sea, in INRA, for receiving me there: Mariam, María José, Julien, Eric, Pierre, Le, Huan, Messa, David, Fatma and all the staff for all their help. -

The Metabolism of Androstenone and Other Steroid Hormone Conjugates in Relation to Boar Taint
The Metabolism of Androstenone and Other Steroid Hormone Conjugates in Relation to Boar Taint by Heidi M. Laderoute A Thesis presented to The University of Guelph In partial fulfillment of requirements for the degree of Master of Science in Animal and Poultry Science with Toxicology Guelph, Ontario, Canada © Heidi M. Laderoute, April, 2015 ABSTRACT THE METABOLISM OF ANDROSTENONE AND OTHER STEROID HORMONE CONJUGATES IN RELATION TO BOAR TAINT Heidi M. Laderoute Advisor: University of Guelph, 2015 Dr. E.J. Squires Increased public interest in the welfare of pigs reared for pork production has led to an increased effort in finding new approaches for controlling the unpleasant odour and flavour from heated pork products known as boar taint. Therefore, this study investigated the metabolism of androstenone and the enzymes involved in its sulfoconjugation in order to further understand the pathways and genes involved in the development of this meat quality defect. Leydig cells that were incubated with androstenone produced 3-keto- sulfoxy-androstenone, providing direct evidence, for the first time, that sulfoconjugation of this steroid does occur in the boar. In addition, human embryonic kidney cells that were overexpressed with porcine sulfotransferase (SULT) enzymes showed that SULT2A1, but not SULT2B1, was responsible for sulfoconjugating androstenone. These findings emphasize the importance of conjugation in steroid metabolism and its relevance to boar taint is discussed. ACKNOWLEDGEMENTS I would like to gratefully and sincerely thank my advisor, Dr. E. James Squires, for providing me with the opportunity to be a graduate student and for introducing me to the world of boar taint. This project would not have been possible without your guidance, encouragement, and patience over the last few years. -

Intratumoral Estrogen Sulfotransferase Induction Contributes to the Anti-Breast Cancer Effects of the Dithiocarbamate Derivative TM208
npg Acta Pharmacologica Sinica (2015) 36: 1246–1255 © 2015 CPS and SIMM All rights reserved 1671-4083/15 www.nature.com/aps Original Article Intratumoral estrogen sulfotransferase induction contributes to the anti-breast cancer effects of the dithiocarbamate derivative TM208 Xi-wei JI 1, 2, Guang-ping CHEN4, Yan SONG5, Ming HUA1, Li-jie WANG1, Liang LI1, Yin YUAN1, Si-yuan WANG1, Tian-yan ZHOU1, 3, *, Wei LU1, 3 1Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China; 2Institute of Clinical Pharmacology, Peking University First Hospital, Beijing 100191, China; 3State Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China; 4Department of Physiological Sciences, Center for Veterinary Health Sciences, Oklahoma State University, Stillwater, OK 74078, USA; 5Department of Molecular and Cellular Pharmacology, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China Aim: Sulfotransferase-catalyzed sulfation is the most important pathway for inactivating estrogens. Thus, activation of estrogen sulfotransferase (EST) may be an alternative approach for the treatment of estrogen-dependent breast cancer. In this study we investigated the involvement of EST in anti-breast cancer effects of the dithiocarbamate derivative TM208 in vitro and in vivo. Methods: The viability of human breast cancer MCF-7 cells was determined using a SBB assay. Nude mice bearing MCF-7 cells were orally administered TM208 (50 and 150 mg·kg-1·d-1) for 18 days. The xenograft tumors and uteri were collected. The mRNA expression of EST was examined with real-time PCR. EST protein was detected with Western blot, ELISA or immunohistochemical staining assays. A radioactive assay was used to measure the EST activity. -

The Role of Estrogen Sulfotransferase in Ischemic Acute Kidney Injury
Title Page The role of estrogen sulfotransferase in ischemic acute kidney injury By Anne Caroline Silva Barbosa Bachelor of Pharmacy, UFRN, 2015 Submitted to the Graduate Faculty of the School of Pharmacy in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2020 i Committee Membership Page UNIVERSITY OF PITTSBURGH SCHOOL OF PHARMACY This dissertation was presented By Anne Caroline Silva Barbosa It was defended on July 17, 2020 And approved by Christian Fernandez, PhD, Assistant Professor, Pharmaceutical Sciences Robert B. Gibbs, PhD, Professor, Pharmaceutical Sciences Youhua Liu, MD, PhD, Professor, Pathology Thomas D. Nolin, PhD, Associate Professor, Pharmacy & Therapeutics Dissertation Advisor: Wen Xie, MD, PhD, Professor, Pharmaceutical Sciences ii Copyright © by Anne Caroline Silva Barbosa 2020 iii Abstract The role of estrogen sulfotransferase in ischemic acute kidney injury Anne Caroline Silva Barbosa, PhD University of Pittsburgh, 2020 Acute kidney injury (AKI) is a sudden impairment of kidney function. It has been suggested that estrogens may protect mice from AKI. Estrogen sulfotransferase (SULT1E1/EST) plays an important role in estrogen homeostasis by sulfonating and deactivating estrogens, but the role of SULT1E1 in AKI has not been previously reported. In this dissertation, Wild-type (WT) mice, Sult1e1 knockout (Sult1e1 KO) mice, and Sult1e1 KO mice with hepatic reconstitution of Sult1e1 were subjected to a bilateral kidney ischemia-reperfusion model of AKI, in the absence or presence of gonadectomy. Kidney injury was assessed at biochemical, histological and gene expression levels. WT mice treated with the Sult1e1 inhibitor triclosan were also used to determine the effect of pharmacological inhibition of Sult1e1 on AKI. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Obesity-Induced Diabetes (Diabesity) in C57BL/Ksj Mice Produces Aberrant Trans-Regulation of Sex Steroid Sulfotransferase Genes
Obesity-induced diabetes (diabesity) in C57BL/KsJ mice produces aberrant trans-regulation of sex steroid sulfotransferase genes. E H Leiter, H D Chapman J Clin Invest. 1994;93(5):2007-2013. https://doi.org/10.1172/JCI117194. Research Article The diabetes (db) gene is a recessive obesity mutation in the mouse capable of producing diabetes only through interaction with heretofore undefined modifiers in the genetic background of certain inbred strains. Here we identify the genetic map locations of androgen and estrogen sulfotransferase genes important in maintaining the balance of active sex steroids in the liver. The Std locus encoding dehydroepiandrosterone sulfotransferase was mapped to proximal Chromosome 7, and the Ste locus encoding estrogen sulfotransferase was mapped to Chromosome 5. The db mutation in the diabetes-susceptible C57BL/KsJ strain aberrantly regulated mRNA transcript levels from these two loci. Hepatic Ste mRNA transcripts were increased from undetectable levels in normal males and females to high levels in db/db mice of both sexes. An anomalous suppression of Std transcription was observed in db/db females, but not in normal females. These reciprocal changes in mRNA concentrations in mutant females were reflected by an induction of a high affinity estrogen sulfotransferase activity and a concomitant loss of dehydroepiandrosterone sulfotransferase activity. These db gene-elicited effects were specific for the sex steroid sulfotransferases since other potential sex steroid metabolizing enzymes (phenol sulfotransferase, sex steroid sulfohydrolase, and UDP-glucuronyltransferase) were unaffected. These aberrant changes would virilize hepatic metabolism in females by increasing the ratio of active androgens to estrogens. In human females, non-insulin-dependent diabetes mellitus often develops when visceral […] Find the latest version: https://jci.me/117194/pdf Obesity-induced Diabetes (Diabesity) in C57BL/KsJ Mice Produces Aberrant trans-Regulation of Sex Steroid Sulfotransferase Genes Edward H. -
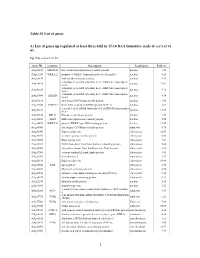
Table S1 List of Genes A) List of Genes Up-Regulated at Least Three-Fold in 15-16 DAA Immature Seeds of Ex1/Ex2 Vs Wt
Table S1 List of genes A) List of genes up-regulated at least three-fold in 15-16 DAA immature seeds of ex1/ex2 vs wt Up (74) ex1ex2 vs wt Gene ID Common Description Localization Fold (+) At2g26150 ATHSFA2 heat shock transcription factor family protein nucleus 3.01 At4g01250 WRKY22 member of WRKY Transcription Factor; Group II-e nucleus 4.24 At1g68670 myb family transcription factor nucleus 5.64 a member of the ERF subfamily B-3 of ERF/AP2 transcription At4g34410 nucleus 11.02 factor a member of the ERF subfamily B-2 of ERF/AP2 transcription At2g47520 nucleus 4.72 factor a member of the ERF subfamily B-3 of ERF/AP2 transcription At4g17490 ATERF6 nucleus 4.99 factor At3g54810 zinc finger (GATA type) family protein, nucleus 4.98 At1g32640 ATMYC2 Basic helix-loop-helix (bHLH) protein (RAP-1) nucleus 3.62 a member of the DREB subfamily A-5 of ERF/AP2 transcription At1g19210 nucleus 17.89 factor At5g59820 RHL41 Encodes a zinc finger protein nucleus 4.55 At2g18160 GBF5 bZIP transcription factor family protein nucleus 5.25 At2g38470 WRKY33 putative WRKY-type DNA binding protein nucleus 17.47 At3g55980 zinc finger (CCCH-type) family protein, unknown 4.70 At3g10040 Expressed protein chloroplast 21.97 At4g10270 wound-responsive family protein chloroplast 8.68 At3g19680 Expressed protein chloroplast 3.63 At3g13310 DNAJ heat shock N-terminal domain-containing protein chloroplast 9.48 At3g52760 integral membrane Yip1 family protein, Yip1 domain chloroplast 3.11 At4g27280 calcium-binding EF hand family protein chloroplast 3.81 At5g26030 ferrochelatase-I -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Xenobiotic Metabolism, Disposition, and Regulation by Receptors: from Biochemical Phenomenon to Predictors of Major Toxicities
TOXICOLOGICAL SCIENCES 120(S1), S49–S75 (2011) doi:10.1093/toxsci/kfq338 Advance Access publication November 8, 2010 Xenobiotic Metabolism, Disposition, and Regulation by Receptors: From Biochemical Phenomenon to Predictors of Major Toxicities Curtis J. Omiecinski,1 John P. Vanden Heuvel, Gary H. Perdew, and Jeffrey M. Peters Department of Veterinary and Biomedical Sciences, Center for Molecular Toxicology and Carcinogenesis, Penn State University, University Park, Pennsylvania 16802 1To whom correspondence should be addressed at Department of Veterinary and Biomedical Sciences, Center for Molecular Toxicology and Carcinogenesis, Downloaded from 101 Life Sciences Building, Penn State University, University Park, PA 16802. Fax: (814) 863-1696. E-mail: [email protected]. Received September 1, 2010; accepted November 1, 2010 processes. Indeed, in the past 50 years, much has been gleaned To commemorate the 50th anniversary of the Society of regarding our mechanistic understanding of chemical toxicities toxsci.oxfordjournals.org Toxicology, this special edition article reviews the history and contributed by these coordinate biotransformation systems. current scope of xenobiotic metabolism and transport, with special From the early discoveries of these pathways, our scientific emphasis on the discoveries and impact of selected ‘‘xenobiotic receptors.’’ This overall research realm has witnessed dynamic perspective has witnessed extraordinary advances led by the development in the past 50 years, and several of the key milestone availability of new -

Specific Estrogen Sulfotransferase (SULT1E1) Substrates and Molecular Imaging Probe Candidates
Specific estrogen sulfotransferase (SULT1E1) substrates and molecular imaging probe candidates Graham B. Colea, Gyochang Keuma, Jie Liua, Gary W. Smallb, Nagichettiar Satyamurthya, Vladimir Kepea, and Jorge R. Barrioa,1 aDepartments of Molecular and Medical Pharmacology and bPsychiatry and Biobehavioral Sciences, David Geffen School of Medicine at University of California, Los Angeles, CA 90095-6948 Communicated by Michael E. Phelps, University of California, Los Angeles, Los Angeles, CA, December 28, 2009 (received for review July 24, 2009) This work focuses on the development of specific substrates for estrogen sulfotransferase (SULT1E1) to produce molecular imaging probes for this enzyme. SULT1E1 is a key enzyme in estrogen homeostasis, playing a central role in the prevention and develop- ment of human disease. In vitro sulfation assays showed alkyl and aryl substitutions to a fused heterocyclic system modeled after β- naphthol (βN), based on compounds that interact with the estrogen receptor, rendered several molecules with enhanced specificity for SULT1E1 over SULT1A1*1, SULT1A1*2, SULT1A3, and SULT2A1. Sev- eral 6-hydroxy-2-arylbenzothiazoles tested demonstrated excel- —V ∕K — K lent affinity max m ratios and specificity for SULT1E1. m values ranged from 0.12–2.36 μM. A strong correlation was ob- served between polarity of the 4′-sustituent on the 2-aryl moiety σ ðV ∕K Þ r ¼ 0 964 Fig. 1. (A) Interplay of estrogen sulfation and estrogen receptor (ER). Tissue (Hammett p) and the log max m ( . ). Substrate sensitiv- levels of E2 are regulated by SULT1E1 and STS, thus, modulating E2 interac- ity is influenced by the acidity of the 6-phenolic group tion with the estrogen receptor (ER). -

(12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur Et Al
US 20150240226A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0240226A1 Mathur et al. (43) Pub. Date: Aug. 27, 2015 (54) NUCLEICACIDS AND PROTEINS AND CI2N 9/16 (2006.01) METHODS FOR MAKING AND USING THEMI CI2N 9/02 (2006.01) CI2N 9/78 (2006.01) (71) Applicant: BP Corporation North America Inc., CI2N 9/12 (2006.01) Naperville, IL (US) CI2N 9/24 (2006.01) CI2O 1/02 (2006.01) (72) Inventors: Eric J. Mathur, San Diego, CA (US); CI2N 9/42 (2006.01) Cathy Chang, San Marcos, CA (US) (52) U.S. Cl. CPC. CI2N 9/88 (2013.01); C12O 1/02 (2013.01); (21) Appl. No.: 14/630,006 CI2O I/04 (2013.01): CI2N 9/80 (2013.01); CI2N 9/241.1 (2013.01); C12N 9/0065 (22) Filed: Feb. 24, 2015 (2013.01); C12N 9/2437 (2013.01); C12N 9/14 Related U.S. Application Data (2013.01); C12N 9/16 (2013.01); C12N 9/0061 (2013.01); C12N 9/78 (2013.01); C12N 9/0071 (62) Division of application No. 13/400,365, filed on Feb. (2013.01); C12N 9/1241 (2013.01): CI2N 20, 2012, now Pat. No. 8,962,800, which is a division 9/2482 (2013.01); C07K 2/00 (2013.01); C12Y of application No. 1 1/817,403, filed on May 7, 2008, 305/01004 (2013.01); C12Y 1 1 1/01016 now Pat. No. 8,119,385, filed as application No. PCT/ (2013.01); C12Y302/01004 (2013.01); C12Y US2006/007642 on Mar. 3, 2006. -

Mediated Activation of Estrogen Sulfotransferase
Research Article Glucocorticoids Antagonize Estrogens by Glucocorticoid Receptor–Mediated Activation of Estrogen Sulfotransferase Haibiao Gong,1 Michael J. Jarzynka,2 Timothy J. Cole,4 Jung Hoon Lee,1 Taira Wada,1 Bin Zhang,1 Jie Gao,1 Wen-Chao Song,5 Donald B. DeFranco,3 Shi-Yuan Cheng,2 and Wen Xie1,3 1Center for Pharmacogenetics and Department of Pharmaceutical Sciences, 2University of Pittsburgh Cancer Institute and Department of Pathology, and 3Department of Pharmacology, University of Pittsburgh, Pittsburgh, Pennsylvania; 4Department of Biochemistry and Molecular Biology, Monash University, Clayton, Victoria, Australia; and 5Institute for Translational Medicine and Therapeutics and Department of Pharmacology, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania Abstract superfamily. In the absence of a ligand, GR resides in the cytoplasm Glucocorticoids and estrogens are two classes of steroid associating with the heat shock protein hsp90 and several other hormones that have essential but distinct physiologic proteins. Upon ligand binding, GR is dissociated with hsp90 and functions. Estrogens also represent a risk factor for breast translocated into the nucleus, where it activates gene expression by cancer. It has been suggested that glucocorticoids can binding to glucocorticoid-responsive elements (GRE) located in the attenuate estrogen responses, but the mechanism by which promoter regions of target genes (4–6). glucocorticoids inhibit estrogenic activity is unknown. In this Estrogens are sex hormones essential for mammals’ reproduc- tion. Although produced primarily in the ovary, estrogens circulate study, we show that activation of glucocorticoid receptor (GR) by dexamethasone (DEX) induced the expression and systemically and exert their effects on various reproductive and activity of estrogen sulfotransferase (SULT1E1 or EST), nonreproductive tissues (7, 8).