Supplementary Material (ESI) for Natural Product Reports
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

KO Kidney.Xlsx
Supplemental Table 18: Dietary Impact on the CGL KO Kidney Sulfhydrome DR/AL Accession Molecular Cysteine Spectral Protein Name Number Alternate ID Weight Residues Count Ratio P‐value Ig gamma‐2A chain C region, A allele P01863 (+1) Ighg 36 kDa 10 C 5.952 0.03767 Heterogeneous nuclear ribonucleoprotein M Q9D0E1 (+1) Hnrnpm 78 kDa 6 C 5.000 0.00595 Phospholipase D3 O35405 Pld3 54 kDa 8 C 4.167 0.04761 Ig kappa chain V‐V region L7 (Fragment) P01642 Gm10881 13 kDa 2 C 2.857 0.01232 UPF0160 protein MYG1, mitochondrial Q9JK81 Myg1 43 kDa 7 C 2.333 0.01613 Copper homeostasis protein cutC homolog Q9D8X1 Cutc 29 kDa 7 C 10.333 0.16419 Corticosteroid‐binding globulin Q06770 Serpina6 45 kDa 3 C 10.333 0.16419 28S ribosomal protein S22, mitochondrial Q9CXW2 Mrps22 41 kDa 2 C 7.333 0.3739 Isoform 3 of Agrin A2ASQ1‐3 Agrn 198 kDa 2 C 7.333 0.3739 3‐oxoacyl‐[acyl‐carrier‐protein] synthase, mitochondrial Q9D404 Oxsm 49 kDa 11 C 7.333 0.3739 Cordon‐bleu protein‐like 1 Q3UMF0 (+3)Cobll1 137 kDa 10 C 5.833 0.10658 ADP‐sugar pyrophosphatase Q9JKX6 Nudt5 24 kDa 5 C 4.167 0.15819 Complement C4‐B P01029 C4b 193 kDa 29 C 3.381 0.23959 Protein‐glutamine gamma‐glutamyltransferase 2 P21981 Tgm2 77 kDa 20 C 3.381 0.23959 Isochorismatase domain‐containing protein 1 Q91V64 Isoc1 32 kDa 5 C 3.333 0.10588 Serpin B8 O08800 Serpinb8 42 kDa 11 C 2.903 0.06902 Heterogeneous nuclear ribonucleoprotein A0 Q9CX86 Hnrnpa0 31 kDa 3 C 2.667 0.5461 Proteasome subunit beta type‐8 P28063 Psmb8 30 kDa 5 C 2.583 0.36848 Ig kappa chain V‐V region MOPC 149 P01636 12 kDa 2 C 2.583 0.36848 -

C19) United States C12) Patent Application Publication C10) Pub
1111111111111111 IIIIII IIIII 1111111111 11111 11111 111111111111111 1111111111 1111111111 11111111 US 20200081016Al c19) United States c12) Patent Application Publication c10) Pub. No.: US 2020/0081016 Al Talaat et al. (43) Pub. Date: Mar. 12, 2020 (54) BIOMARKERS FOR EARLY DIAGNOSIS Publication Classification AND DIFFERENTIATION OF (51) Int. Cl. MYCOBACTERIAL INFECTION GOIN 33/68 (2006.01) C12Q 116851 (2006.01) (71) Applicant: Wisconsin Alumni Research GOIN 33/569 (2006.01) Foundation, Madison, WI (US) (52) U.S. Cl. (72) Inventors: Adel Mohamed Talaat, Madison, WI CPC ......... GOIN 33/6854 (2013.01); GOIN 33/68 (US); Chia-wei Wu, Madison, WI (US) (2013.01); GOIN 2800/50 (2013.01); GOIN 33/5695 (2013.01); GOIN 2800/26 (2013.01); (21) Appl. No.: 16/555,819 C12Q 116851 (2013.01) (22) Filed: Aug. 29, 2019 (57) ABSTRACT Mycobacterial-specific biomarkers and methods of using Related U.S. Application Data such biomarkers for diagnosis of mycobacterial infection in (60) Provisional application No. 62/728,387, filed on Sep. a mammal are disclosed. 7, 2018. Specification includes a Sequence Listing. Patent Application Publication Mar. 12, 2020 Sheet 1 of 10 US 2020/0081016 Al FIG. 1 ·~{:: -{t i * !lpNbiNi$ 1 !lpN p~ra 111:111111 llillllll: 111!11,111llltllllll~ 11111 ■111 ~; C,,Nmnsus KR.IGINMTKX L.lC(X.AXXXXG AXXXXMPXTX RXO-GXVXXVG VKVXPWIPTX ® • ® l I I iipN lK>V(S ~Hl!lli!Wiofflij 1!11.llofJiillj mllB~lijftlt flol=fiolill ••t-il-~MM ~9 llpN p~ra HfHJoffit:torti ilffllGNillm miJllt~ttiollf ~•01:101111 llm:l:l1IA@~ iOO C,,nstmsus XXRXLXXGRS Vt IOGNT.LDP i LOt.MLSXXR XXGXOG.I...XVO ODXXXSR:AXM t2:;: i-/4~~ ! l 1 I~~~~b;:: llllil~l:1:1 llil 111111:1~:111~ 1111111::;1 1lllilllll: ~:~ C,,nimnsus XXXXXXXPGP QtHVDVXXI...X XPGPAGXIPA RHYRPXGGXX QXPt.l...VFYHG Consl:lrvat,ofl -:§;::. -

Structural Insights Into Histone Modifying Enzymes
Wayne State University Wayne State University Theses January 2019 Structural Insights Into Histone Modifying Enzymes Shruti Amle Wayne State University, [email protected] Follow this and additional works at: https://digitalcommons.wayne.edu/oa_theses Part of the Biochemistry Commons, and the Molecular Biology Commons Recommended Citation Amle, Shruti, "Structural Insights Into Histone Modifying Enzymes" (2019). Wayne State University Theses. 693. https://digitalcommons.wayne.edu/oa_theses/693 This Open Access Embargo is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Theses by an authorized administrator of DigitalCommons@WayneState. STRUCTURAL INSIGHTS INTO HISTONE MODIFYING ENZYMES by SHRUTI AMLE THESIS Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE 2019 MAJOR: BIOCHEMISTRY AND MOLECULAR BIOLOGY Approved By: _________________________________________ Advisor Date _________________________________________ _________________________________________ _________________________________________ i ACKNOWLEDGEMENTS Writing this thesis has been extremely captivating and gratifying. I take this opportunity to express my deep and sincere acknowledgements to the number of people for extending their generous support and unstinted help during my entire study. Firstly, I would like to express my respectful regards and deep sense of gratitude to my advisor, Dr. Zhe Yang. I am extremely honored to study and work under his guidance. His vision, ideals, timely motivation and immense knowledge had a deep influence on my entire journey of this career. Without his understanding and support, it would not have been possible to complete this research successfully. I also owe my special thanks to my committee members: Dr. -

(12) Patent Application Publication (10) Pub. No.: US 2013/0089535 A1 Yamashiro Et Al
US 2013 0089535A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0089535 A1 Yamashiro et al. (43) Pub. Date: Apr. 11, 2013 (54) AGENT FOR REDUCING ACETALDEHYDE Publication Classification NORAL CAVITY (51) Int. Cl. (75) Inventors: Kan Yamashiro, Kakamigahara-shi (JP); A68/66 (2006.01) Takahumi Koyama, Kakamigahara-shi A638/51 (2006.01) (JP) A61O 11/00 (2006.01) A638/44 (2006.01) Assignee: AMANOENZYME INC., Nagoya-shi (52) U.S. Cl. (73) CPC. A61K 8/66 (2013.01); A61K 38/44 (2013.01); (JP) A61 K38/51 (2013.01); A61O II/00 (2013.01) (21) Appl. No.: 13/703,451 USPC .......... 424/94.4; 424/94.5; 435/191: 435/232 (22) PCT Fled: Jun. 7, 2011 (57) ABSTRACT Disclosed herein is a novel enzymatic agent effective in (86) PCT NO.: PCT/UP2011/062991 reducing acetaldehyde in the oral cavity. It has been found S371 (c)(1), that an aldehyde dehydrogenase derived from a microorgan (2), (4) Date: Dec. 11, 2012 ism belonging to the genus Saccharomyces and a threonine aldolase derived from Escherichia coli are effective in reduc (30) Foreign Application Priority Data ing low concentrations of acetaldehyde. Therefore, an agent for reducing acetaldehyde in the oral cavity is provided, Jun. 19, 2010 (JP) ................................. 2010-140O26 which contains these enzymes as active ingredients. Patent Application Publication Apr. 11, 2013 Sheet 1 of 2 US 2013/0089535 A1 FIG 1) 10.5 1 0 9.9.5 8. 5 CONTROL TA AD (BSA) ENZYME Patent Application Publication Apr. 11, 2013 Sheet 2 of 2 US 2013/0089535 A1 FIG 2) 110 the CONTROL (BSA) 100 354. -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Structural Properties of the Nickel Ions in Urease: Novel Insights Into the Catalytic and Inhibition Mechanisms
Coordination Chemistry Reviews 190–192 (1999) 331–355 www.elsevier.com/locate/ccr Structural properties of the nickel ions in urease: novel insights into the catalytic and inhibition mechanisms Stefano Ciurli a,*, Stefano Benini b, Wojciech R. Rypniewski b, Keith S. Wilson c, Silvia Miletti a, Stefano Mangani d a Institute of Agricultural Chemistry, Uni6ersity of Bologna, Viale Berti Pichat 10, I-40127 Bologna, Italy b European Molecular Biology Laboratory, c/o DESY, Notkestraße 85, D-22603 Hamburg, Germany c Department of Chemistry, Uni6ersity of York, Heslington, York YO15DD, UK d Department of Chemistry, Uni6ersity of Siena, Pian dei Mantellini 44, I-53100 Siena, Italy Accepted 13 March 1999 Contents Abstract.................................................... 331 1. Biological background ......................................... 332 2. Spectroscopic investigations of the urease active site structure .................. 333 3. Crystallographic studies of the native enzyme ............................ 334 4. Crystallographic studies of urease mutants.............................. 341 5. Crystallographic studies of urease–inhibitor complexes ...................... 345 6. Crystallographic study of a transition state analogue bound to urease.............. 348 7. A novel proposal for the urease mechanism ............................. 350 References .................................................. 353 Abstract This work provides a comprehensive critical summary of urease spectroscopy, crystallogra- phy, inhibitor binding, and site-directed -

ADAMTS13 and 15 Are Not Regulated by the Full Length and N‑Terminal Domain Forms of TIMP‑1, ‑2, ‑3 and ‑4
BIOMEDICAL REPORTS 4: 73-78, 2016 ADAMTS13 and 15 are not regulated by the full length and N‑terminal domain forms of TIMP‑1, ‑2, ‑3 and ‑4 CENQI GUO, ANASTASIA TSIGKOU and MENG HUEE LEE Department of Biological Sciences, Xian Jiaotong-Liverpool University, Suzhou, Jiangsu 215123, P.R. China Received June 29, 2015; Accepted July 15, 2015 DOI: 10.3892/br.2015.535 Abstract. A disintegrin and metalloproteinase with thom- proteolysis activities associated with arthritis, morphogenesis, bospondin motifs (ADAMTS) 13 and 15 are secreted zinc angiogenesis and even ovulation [as reviewed previously (1,2)]. proteinases involved in the turnover of von Willebrand factor Also known as the VWF-cleaving protease, ADAMTS13 and cancer suppression. In the present study, ADAMTS13 is noted for its ability in cleaving and reducing the size of the and 15 were subjected to inhibition studies with the full-length ultra-large (UL) form of the VWF. Reduction in ADAMTS13 and N-terminal domain forms of tissue inhibitor of metallo- activity from either hereditary or acquired deficiency causes proteinases (TIMPs)-1 to -4. TIMPs have no ability to inhibit accumulation of UL-VWF multimers, platelet aggregation and the ADAMTS proteinases in the full-length or N-terminal arterial thrombosis that leads to fatal thrombotic thrombocy- domain form. While ADAMTS13 is also not sensitive to the topenic purpura [as reviewed previously (1,3)]. By contrast, hydroxamate inhibitors, batimastat and ilomastat, ADAMTS15 ADAMTS15 is a potential tumor suppressor. Only a limited app can be effectively inhibited by batimastat (Ki 299 nM). In number of in-depth investigations have been carried out on the conclusion, the present results indicate that TIMPs are not the enzyme; however, expression and profiling studies have shown regulators of these two ADAMTS proteinases. -
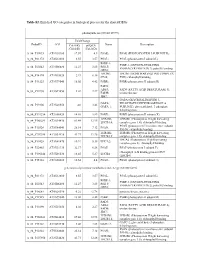
Table S2. Enriched GO Categories in Biological Process for the Shared Degs
Table S2. Enriched GO categories in biological process for the shared DEGs photosynthesis (GO ID:15979) Fold Change ProbeID AGI Col-0(R) pifQ(D) Name Description /Col-0(D) /Col-0(D) A_84_P19035 AT1G30380 17.07 4.9 PSAK; PSAK (PHOTOSYSTEM I SUBUNIT K) A_84_P21372 AT4G12800 8.55 3.57 PSAL; PSAL (photosystem I subunit L) PSBP-1; PSBP-1 (OXYGEN-EVOLVING A_84_P20343 AT1G06680 12.27 3.85 PSII-P; ENHANCER PROTEIN 2); poly(U) binding OEE2; LHCB6; LHCB6 (LIGHT HARVESTING COMPLEX A_84_P14174 AT1G15820 23.9 6.16 CP24; PSII); chlorophyll binding A_84_P11525 AT1G79040 16.02 4.42 PSBR; PSBR (photosystem II subunit R) FAD5; ADS3; FAD5 (FATTY ACID DESATURASE 5); A_84_P19290 AT3G15850 4.02 2.27 FADB; oxidoreductase JB67; GAPA (GLYCERALDEHYDE 3- GAPA; PHOSPHATE DEHYDROGENASE A A_84_P19306 AT3G26650 4.6 3.43 GAPA-1; SUBUNIT); glyceraldehyde-3-phosphate dehydrogenase A_84_P193234 AT2G06520 14.01 3.89 PSBX; PSBX (photosystem II subunit X) LHB1B1; LHB1B1 (Photosystem II light harvesting A_84_P160283 AT2G34430 89.44 32.95 LHCB1.4; complex gene 1.4); chlorophyll binding PSAN (photosystem I reaction center subunit A_84_P10324 AT5G64040 26.14 7.12 PSAN; PSI-N); calmodulin binding LHB1B2; LHB1B2 (Photosystem II light harvesting A_84_P207958 AT2G34420 41.71 12.26 LHCB1.5; complex gene 1.5); chlorophyll binding LHCA2 (Photosystem I light harvesting A_84_P19428 AT3G61470 10.91 5.36 LHCA2; complex gene 2); chlorophyll binding A_84_P22465 AT1G31330 32.37 6.58 PSAF; PSAF (photosystem I subunit F) chlorophyll A-B binding protein CP29 A_84_P190244 AT5G01530 16.45 5.27 LHCB4 -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Serine Proteases with Altered Sensitivity to Activity-Modulating
(19) & (11) EP 2 045 321 A2 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 08.04.2009 Bulletin 2009/15 C12N 9/00 (2006.01) C12N 15/00 (2006.01) C12Q 1/37 (2006.01) (21) Application number: 09150549.5 (22) Date of filing: 26.05.2006 (84) Designated Contracting States: • Haupts, Ulrich AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 51519 Odenthal (DE) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • Coco, Wayne SK TR 50737 Köln (DE) •Tebbe, Jan (30) Priority: 27.05.2005 EP 05104543 50733 Köln (DE) • Votsmeier, Christian (62) Document number(s) of the earlier application(s) in 50259 Pulheim (DE) accordance with Art. 76 EPC: • Scheidig, Andreas 06763303.2 / 1 883 696 50823 Köln (DE) (71) Applicant: Direvo Biotech AG (74) Representative: von Kreisler Selting Werner 50829 Köln (DE) Patentanwälte P.O. Box 10 22 41 (72) Inventors: 50462 Köln (DE) • Koltermann, André 82057 Icking (DE) Remarks: • Kettling, Ulrich This application was filed on 14-01-2009 as a 81477 München (DE) divisional application to the application mentioned under INID code 62. (54) Serine proteases with altered sensitivity to activity-modulating substances (57) The present invention provides variants of ser- screening of the library in the presence of one or several ine proteases of the S1 class with altered sensitivity to activity-modulating substances, selection of variants with one or more activity-modulating substances. A method altered sensitivity to one or several activity-modulating for the generation of such proteases is disclosed, com- substances and isolation of those polynucleotide se- prising the provision of a protease library encoding poly- quences that encode for the selected variants.