ERK5 Is Activated by Oncogenic BRAF and Promotes Melanoma Growth
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Influencers on Thyroid Cancer Onset: Molecular Genetic Basis
G C A T T A C G G C A T genes Review Influencers on Thyroid Cancer Onset: Molecular Genetic Basis Berta Luzón-Toro 1,2, Raquel María Fernández 1,2, Leticia Villalba-Benito 1,2, Ana Torroglosa 1,2, Guillermo Antiñolo 1,2 and Salud Borrego 1,2,* 1 Department of Maternofetal Medicine, Genetics and Reproduction, Institute of Biomedicine of Seville (IBIS), University Hospital Virgen del Rocío/CSIC/University of Seville, 41013 Seville, Spain; [email protected] (B.L.-T.); [email protected] (R.M.F.); [email protected] (L.V.-B.); [email protected] (A.T.); [email protected] (G.A.) 2 Centre for Biomedical Network Research on Rare Diseases (CIBERER), 41013 Seville, Spain * Correspondence: [email protected]; Tel.: +34-955-012641 Received: 3 September 2019; Accepted: 6 November 2019; Published: 8 November 2019 Abstract: Thyroid cancer, a cancerous tumor or growth located within the thyroid gland, is the most common endocrine cancer. It is one of the few cancers whereby incidence rates have increased in recent years. It occurs in all age groups, from children through to seniors. Most studies are focused on dissecting its genetic basis, since our current knowledge of the genetic background of the different forms of thyroid cancer is far from complete, which poses a challenge for diagnosis and prognosis of the disease. In this review, we describe prevailing advances and update our understanding of the molecular genetics of thyroid cancer, focusing on the main genes related with the pathology, including the different noncoding RNAs associated with the disease. -

Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC)
cancers Review Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC) Chiara Diquigiovanni * and Elena Bonora Unit of Medical Genetics, Department of Medical and Surgical Sciences, University of Bologna, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-208-8418 Simple Summary: Non-medullary thyroid carcinoma (NMTC) originates from thyroid follicular epithelial cells and is considered familial when occurs in two or more first-degree relatives of the patient, in the absence of predisposing environmental factors. Familial NMTC (FNMTC) cases show a high genetic heterogeneity, thus impairing the identification of pivotal molecular changes. In the past years, linkage-based approaches identified several susceptibility loci and variants associated with NMTC risk, however only few genes have been identified. The advent of next-generation sequencing technologies has improved the discovery of new predisposing genes. In this review we report the most significant genes where variants predispose to FNMTC, with the perspective that the integration of these new molecular findings in the clinical data of patients might allow an early detection and tailored therapy of the disease, optimizing patient management. Abstract: Non-medullary thyroid carcinoma (NMTC) is the most frequent endocrine tumor and originates from the follicular epithelial cells of the thyroid. Familial NMTC (FNMTC) has been defined in pedigrees where two or more first-degree relatives of the patient present the disease in absence of other predisposing environmental factors. Compared to sporadic cases, FNMTCs are often multifocal, recurring more frequently and showing an early age at onset with a worse outcome. FNMTC cases Citation: Diquigiovanni, C.; Bonora, E. -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

The MKKK62-MKK3-MAPK7/14 Module Negatively Regulates Seed
Mao et al. Rice (2019) 12:2 https://doi.org/10.1186/s12284-018-0260-z ORIGINAL ARTICLE Open Access The MKKK62-MKK3-MAPK7/14 module negatively regulates seed dormancy in rice Xingxue Mao1,2†, Jianjun Zhang3†, Wuge Liu1,2†, Shijuan Yan4, Qing Liu1,2, Hua Fu1,2, Junliang Zhao1,2, Wenjie Huang4, Jingfang Dong1,2, Shaohong Zhang1,2, Tifeng Yang1,2, Wu Yang1,2, Bin Liu1,2* and Feng Wang1,2* Abstract Background: Seed dormancy directly affects the phenotype of pre-harvest sprouting, and ultimately affects the quality and yield of rice seeds. Although many genes controlling seed dormancy have been cloned from cereals, the regulatory mechanisms controlling this process are complex, and much remains unknown. The MAPK cascade is involved in many signal transduction pathways. Recently, MKK3 has been reported to be involved in the regulation of seed dormancy, but its mechanism of action is unclear. Results: We found that MKKK62-overexpressing rice lines (OE) lost seed dormancy. Further analyses showed that the abscisic acid (ABA) sensitivity of OE lines was decreased. In yeast two-hybrid experiments, MKKK62 interacted with MKK3, and MKK3 interacted with MAPK7 and MAPK14. Knock-out experiments confirmed that MKK3, MAPK7, and MAPK14 were involved in the regulation of seed dormancy. The OE lines showed decreased transcript levels of OsMFT, a homolog of a gene that controls seed dormancy in wheat. The up-regulation of OsMFT in MKK3-knockout lines (OE/mkk3) and MAPK7/14-knockout lines (OE/mapk7/mapk14) indicated that the MKKK62-MKK3-MAPK7/ MAPK14 system controlled seed dormancy by regulating the transcription of OsMFT. -

Molecular Mechanisms Underlying Innate Immune Kinase
MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION APPROVED BY SUPERVISORY COMMITTEE Michael A White, Ph.D. Melanie H. Cobb, Ph.D. Lawrence Lum, Ph.D. John D. Minna, M.D. DEDICATION This work is dedicated to my mother and Arlene for their love and support. ACKNOWLEDGEMENTS I am very grateful to my mentor, Dr. Michael White, for his continuous support and guidance through the entire study. I really appreciate his inspiration, patience, and generosity. I would also like to thank my committee members, Dr. Cobb, Dr. Lum, and Dr. Minna, for their invaluable advice and discussion. I thank all the White lab members and my friends for their help, suggestion, and discussion. Particularly, I would like to thank Rosie, Michael, Brian, Tzuling, and Malia for their long-term support and collaboration. I would also like to thank my friends, Veleka, Pei-Ling, Jen-Chieh, Shu-Yi, Chih-Chiang, Jen-Shuan, Yi-Chun, and Yu-San for their friendship. I am grateful to Drs. Rolf Brekken, Zhijian James Chen, Xuetao Cao, Philip Tsichlis, Charles Yeaman, William Hahn, Keqiang Ye, and Shu-Chan Hsu, Bing Su, Dos Sarbassov, Mark Magnuson, David Sabatini, Thomas Tan, and Bert Vogelstein for many of the reagents used in these studies. Finally, and most importantly, I would like to thank my mother and Arlene for their unending support and encouragement. MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION by YI-HUNG OU DISSERTATION Presented to the Faculty of the Graduate School of Biomedical Sciences The University of Texas Southwestern Medical Center at Dallas In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY The University of Texas Southwestern Medical Center at Dallas Dallas, Texas April, 2013 Copyright by YI-HUNG OU, 2013 All Rights Reserved MOLECULAR MECHANISMS UNDERLYING INNATE IMMUNE KINASE TBK1-DRIVEN ONCOGENIC TRANSFORMATION Publication No. -
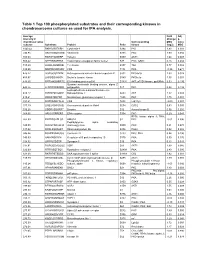
Table 1 Top 100 Phosphorylated Substrates and Their Corresponding Kinases in Chondrosarcoma Cultures As Used for IPA Analysis
Table 1 Top 100 phosphorylated substrates and their corresponding kinases in chondrosarcoma cultures as used for IPA analysis. Average Fold Adj intensity in Change p- chondrosarcoma Corresponding MSC value cultures Substrate Protein Psite kinase (log2) MSC 1043.42 RKKKVSSTKRH Cytohesin-1 S394 PKC 1.83 0.001 746.95 RKGYRSQRGHS Vitronectin S381 PKC 1.00 0.056 709.03 RARSTSLNERP Tuberin S939 AKT1 1.64 0.008 559.42 SPPRSSLRRSS Transcription elongation factor A-like1 S37 PKC; GSK3 0.18 0.684 515.29 LRRSLSRSMSQ Telethonin S157 Titin 0.77 0.082 510.00 MQPDNSSDSDY CD5 T434 PKA -0.35 0.671 476.27 GGRGGSRARNL Heterogeneous nuclear ribonucleoprotein K S302 PKCdelta 1.03 0.028 455.97 LKPGSSHRKTK Bruton's tyrosine kinase S180 PKCbeta 1.55 0.001 444.65 RRRMASMQRTG E1A binding protein p300 S1834 AKT; p70S6 kinase; pp90Rsk 0.53 0.195 Guanine nucleotide binding protein, alpha Z 440.26 HLRSESQRQRR polypeptide S27 PKC 0.88 0.199 6-phosphofructo-2-kinase/fructose-2,6- 424.12 RPRNYSVGSRP biphosphatase 2 S483 AKT 1.32 0.003 419.61 KKKIATRKPRF Metabotropic glutamate receptor 1 T695 PKC 1.75 0.001 391.21 DNSSDSDYDLH CD5 T453 Lck; Fyn -2.09 0.001 377.39 LRQLRSPRRAQ Ras associated protein Rab4 S204 CDC2 0.63 0.091 376.28 SSQRVSSYRRT Desmin S12 Aurora kinase B 0.56 0.255 369.05 ARIGGSRRERS EP4 receptor S354 PKC 0.29 0.543 RPS6 kinase alpha 3; PKA; 367.99 EPKRRSARLSA HMG14 S7 PKC -0.01 0.996 Peptidylglycine alpha amidating 349.08 SRKGYSRKGFD monooxygenase S930 PKC 0.21 0.678 347.92 RRRLSSLRAST Ribosomal protein S6 S236 PAK2 0.02 0.985 346.84 RSNPPSRKGSG Connexin -

Members of the Competence Network for Congenital Heart Defects, Germany
Members of the Competence Network for Congenital Heart Defects, Germany Hashim Abdul-Khaliq, Hans-Heiner Kramer, Felix Berger, Brigitte Stiller, Ulrike Bauer, Thomas Pickardt, Sabine Klaassen Family 49 Family 62 Family 226 Family 333 * * * * * * * * TOF AVS ASD ASD PAPVR TOF * * * * ASD PAPVD COA ASD * Family 346 Family 398 VSD * Family 489 * * VSD VSD * * VSD HCM * * * Family 545 BAV AVS ASD PDA Sv AS VSD Family 576 Family 645 Septal defect * * * COA AVS ASD BAV VSD * * * * BAV AVS AVS ASD BAV Family 702 * VSD VSD Family 732 * Family 720 AVSD * * * VSD * * Family 831 TOF TOF TOF Vring TGA VSD * * * * PDA VSD AVS * PDA * * * VSD BAV PFO ASD ASD2 PDA PDA COA Family 1117 Family 1121 * PVS ? * ASD VSD * * * TOF VSD ASD VSD Family 1319 Family 1151 * * * * 2 * ASD ASD * * ASD ASD * * EbA AVS AVS Family 1364 Family 1560 Family 1575 * * TOF VSD ASD VSD infPS TOF * * * VSD PVS PVS PVS VSD * VSD PVS Family 1710 ASD Family 1722 * ASD CHD * VSD VSD * COA TGA DCM ASD BAV SV, MVA COA PVS * DCM DCM ASD HLHS COA BAV Family 2077 Family 2261 * COA, BAV * * * PVS PVS PVS * * infPS infPS BAV ASD * * AVS ASD ASD BAV BAV Family 3500 Family 2558 Family 3315 * * * AVS VSD AVR * * HLHS COA PDA ASD BAV * PVA Family 3501 VSD Family 3503 ? ? * * * VSD HRHS EbA * 3 PVA PAA ASD PDA VSD Family 3505 Family 3540 * * BAV ASD AVS * * * HLHS HLHS TAPVR BAV COA Figure S1. Pedigrees of 32 Danish multiplex CHD families. Circles: females. Squares: males. White symbols: unaffected family members. Filled symbols: affected family members. Triangles: abortion. -

Mutational Profiling of Lipomas
bioRxiv preprint doi: https://doi.org/10.1101/585307; this version posted March 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Mutational profiling of lipomas Deepika Kanojia1*, Pushkar Dakle1, Anand Mayakonda1,2, Rajeev Parameswaran3, Mark E Puhaindran4, Victor Lee Kwan Min5, Vikas Madan1 and H Phillip Koeffler1,6,7 1Cancer Science Institute of Singapore, National University of Singapore, Singapore; 2Epigenomics and Cancer Risk Factors, German Cancer Research Center (DKFZ), Heidelberg, Germany; 3Division of Surgical Oncology, National University Cancer Institute, Singapore; 4Department of Hand and Reconstructive Microsurgery, National University Hospital; 5Department of Pathology, National University Hospital; 6Division of Hematology/Oncology, Cedars-Sinai Medical Center, University of California, School of Medicine, Los Angeles, California, USA; 7National University Cancer Institute, National University Hospital, Singapore. *Correspondence: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/585307; this version posted March 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Lipomas are benign fatty tumors with a high prevalence rate, mostly found in adults but have a good prognosis. Until now, reason for lipoma occurrence not been identified. We performed whole exome sequencing to define the mutational spectrum in ten lipoma patients along with their matching control samples. -

Targeting Mir128-3P Alleviates Myocardial Insulin Resistance And
RESEARCH ARTICLE Targeting mir128-3p alleviates myocardial insulin resistance and prevents ischemia- induced heart failure Andrea Ruiz-Velasco1, Min Zi1, Susanne S Hille2,3, Tayyiba Azam1, Namrita Kaur1, Juwei Jiang1, Binh Nguyen1, Karolina Sekeres4, Pablo Binder1, Lucy Collins1, Fay Pu5, Han Xiao6, Kaomei Guan4, Norbert Frey2,3, Elizabeth J Cartwright1, Oliver J Mu¨ ller2,3*, Xin Wang1*, Wei Liu1* 1Faculty of Biology, Medicine, and Health, the University of Manchester, Manchester, United Kingdom; 2Department of Internal Medicine III, University of Kiel, Kiel, Germany; 3DZHK, German Centre for Cardiovascular Research, Partner Site Hamburg/Kiel/Lu¨ beck, Kiel, Germany; 4Institute of Pharmacology and Toxicology, Faculty of Medicine Carl Gustav Carus, Technische Universitaet Dresden, Dresden, Germany; 5Edinburgh University Medical School, Edinburgh, United Kingdom; 6Institute of Vascular Medicine, Peking University, Beijing, China Abstract Myocardial insulin resistance contributes to heart failure in response to pathological stresses, therefore, a therapeutic strategy to maintain cardiac insulin pathways requires further investigation. We demonstrated that insulin receptor substrate 1 (IRS1) was reduced in failing mouse hearts post-myocardial infarction (MI) and failing human hearts. The mice manifesting severe cardiac dysfunction post-MI displayed elevated mir128-3p in the myocardium. Ischemia- upregulated mir128-3p promoted Irs1 degradation. Using rat cardiomyocytes and human-induced pluripotent stem cell-derived cardiomyocytes, we elucidated that mitogen-activated protein kinase *For correspondence: 7 (MAPK7, also known as ERK5)-mediated CCAAT/enhancer-binding protein beta (CEBPb) [email protected] (OJM); transcriptionally represses mir128-3p under hypoxia. Therapeutically, functional studies [email protected] (XW); demonstrated gene therapy-delivered cardiac-specific MAPK7 restoration or overexpression of [email protected] (WL) CEBPb impeded cardiac injury after MI, at least partly due to normalization of mir128-3p. -

BMK1 Kinase Suppresses Epithelial–Mesenchymal Transition Through the Akt/Gsk3b Signaling Pathway
Published OnlineFirst January 26, 2012; DOI: 10.1158/0008-5472.CAN-11-2055 Cancer Tumor and Stem Cell Biology Research BMK1 Kinase Suppresses Epithelial–Mesenchymal Transition through the Akt/GSK3b Signaling Pathway Runqiang Chen, Qingkai Yang, and Jiing-Dwan Lee Abstract Epithelial–mesenchymal transition (EMT) plays a crucial role in the development of cancer metastasis. The – jun mitogen-activated protein (MAP) kinases extracellular signal regulated kinase, c- -NH2-kinase, and p38 have been implicated in promoting EMT, but a role for the MAP kinase BMK1 has not been studied. Here, we report that BMK1 signaling suppresses EMT. BMK1 elevation augmented E-cadherin–mediated cell–cell adhesion, downregulated mesenchymal markers, and decreased cell motility. Conversely, BMK1 silencing attenuated E-cadherin–mediated cell–cell adhesion, upregulated mesenchymal markers, and stimulated cell motility. BMK1 depletion dramatically increased the accumulation of endogenous Snail in the nuclear compartment. Snail accumulation was mediated by Akt/GSK3b signaling, which was activated by a modulation in the expression of the mTOR inhibitor DEPTOR. In support of these observations, BMK1 depletion promoted metastasis in vivo. Together, our findings reveal a novel mechanism of EMT control via mTOR/Akt inhibition that suppresses cancer metastasis. Cancer Res; 72(6); 1–9. Ó2012 AACR. Introduction that control cancer progression. The majority of mitogenic/ – The process of epithelial–mesenchymal transition (EMT) oncogenic signal activated signaling pathways stimulate is critically involved in the progression of human diseases, EMT(2).Inparticular,3ofthe4mitogen-activatedprotein such as cancer metastasis and fibrosis (1). EMT involves (MAP) kinase pathways described to date (namely, Erk, JNK, profound phenotypic changes that include loss of cell–cell and p38; ref. -

Inhibition of ERK 1/2 Kinases Prevents Tendon Matrix Breakdown Ulrich Blache1,2,3, Stefania L
www.nature.com/scientificreports OPEN Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown Ulrich Blache1,2,3, Stefania L. Wunderli1,2,3, Amro A. Hussien1,2, Tino Stauber1,2, Gabriel Flückiger1,2, Maja Bollhalder1,2, Barbara Niederöst1,2, Sandro F. Fucentese1 & Jess G. Snedeker1,2* Tendon extracellular matrix (ECM) mechanical unloading results in tissue degradation and breakdown, with niche-dependent cellular stress directing proteolytic degradation of tendon. Here, we show that the extracellular-signal regulated kinase (ERK) pathway is central in tendon degradation of load-deprived tissue explants. We show that ERK 1/2 are highly phosphorylated in mechanically unloaded tendon fascicles in a vascular niche-dependent manner. Pharmacological inhibition of ERK 1/2 abolishes the induction of ECM catabolic gene expression (MMPs) and fully prevents loss of mechanical properties. Moreover, ERK 1/2 inhibition in unloaded tendon fascicles suppresses features of pathological tissue remodeling such as collagen type 3 matrix switch and the induction of the pro-fbrotic cytokine interleukin 11. This work demonstrates ERK signaling as a central checkpoint to trigger tendon matrix degradation and remodeling using load-deprived tissue explants. Tendon is a musculoskeletal tissue that transmits muscle force to bone. To accomplish its biomechanical function, tendon tissues adopt a specialized extracellular matrix (ECM) structure1. Te load-bearing tendon compart- ment consists of highly aligned collagen-rich fascicles that are interspersed with tendon stromal cells. Tendon is a mechanosensitive tissue whereby physiological mechanical loading is vital for maintaining tendon archi- tecture and homeostasis2. Mechanical unloading of the tissue, for instance following tendon rupture or more localized micro trauma, leads to proteolytic breakdown of the tissue with severe deterioration of both structural and mechanical properties3–5.