Identification of Active Sites in Amidase: Evolutionary Relationship Between
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -
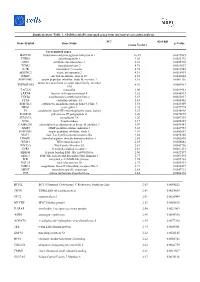
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Biochemical Characterization and Comparison of Aspartylglucosaminidases Secreted in Venom of the Parasitoid Wasps Asobara Tabida and Leptopilina Heterotoma
RESEARCH ARTICLE Biochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma Quentin Coulette1, SeÂverine Lemauf2, Dominique Colinet2, Geneviève PreÂvost1, Caroline Anselme1, Marylène Poirie 2, Jean-Luc Gatti2* a1111111111 1 Unite ªEcologie et Dynamique des Systèmes AnthropiseÂsº (EDYSAN, FRE 3498 CNRS-UPJV), Universite de Picardie Jules Verne, Amiens, France, 2 Universite CoÃte d'Azur, INRA, CNRS, ISA, Sophia Antipolis, a1111111111 France a1111111111 a1111111111 * [email protected] a1111111111 Abstract Aspartylglucosaminidase (AGA) is a low-abundance intracellular enzyme that plays a key OPEN ACCESS role in the last stage of glycoproteins degradation, and whose deficiency leads to human Citation: Coulette Q, Lemauf S, Colinet D, PreÂvost aspartylglucosaminuria, a lysosomal storage disease. Surprisingly, high amounts of AGA- G, Anselme C, Poirie M, et al. (2017) Biochemical characterization and comparison of like proteins are secreted in the venom of two phylogenetically distant hymenopteran para- aspartylglucosaminidases secreted in venom of the sitoid wasp species, Asobara tabida (Braconidae) and Leptopilina heterotoma (Cynipidae). parasitoid wasps Asobara tabida and Leptopilina These venom AGAs have a similar domain organization as mammalian AGAs. They share heterotoma. PLoS ONE 12(7): e0181940. https:// with them key residues for autocatalysis and activity, and the mature - and -subunits also doi.org/10.1371/journal.pone.0181940 α β form an (αβ)2 structure in solution. Interestingly, only one of these AGAs subunits (α for Editor: Erjun Ling, Institute of Plant Physiology and AtAGA and for LhAGA) is glycosylated instead of the two subunits for lysosomal human Ecology Shanghai Institutes for Biological β Sciences, CHINA AGA (hAGA), and these glycosylations are partially resistant to PGNase F treatment. -

Biosynthesis of Natural Products Containing Β-Amino Acids
Natural Product Reports Biosynthesis of natural products containing β -amino acids Journal: Natural Product Reports Manuscript ID: NP-REV-01-2014-000007.R1 Article Type: Review Article Date Submitted by the Author: 21-Apr-2014 Complete List of Authors: Kudo, Fumitaka; Tokyo Institute Of Technology, Department of Chemistry Miyanaga, Akimasa; Tokyo Institute Of Technology, Department of Chemistry Eguchi, T; Tokyo Institute Of Technology, Department of Chemistry and Materials Science Page 1 of 20 Natural Product Reports NPR RSC Publishing REVIEW Biosynthesis of natural products containing βββ- amino acids Cite this: DOI: 10.1039/x0xx00000x Fumitaka Kudo, a Akimasa Miyanaga, a and Tadashi Eguchi *b Received 00th January 2014, We focus here on β-amino acids as components of complex natural products because the presence of β-amino acids Accepted 00th January 2014 produces structural diversity in natural products and provides characteristic architectures beyond that of ordinary DOI: 10.1039/x0xx00000x α-L-amino acids, thus generating significant and unique biological functions in nature. In this review, we first survey the known bioactive β-amino acid-containing natural products including nonribosomal peptides, www.rsc.org/ macrolactam polyketides, and nucleoside-β-amino acid hybrids. Next, the biosynthetic enzymes that form β-amino acids from α-amino acids and de novo synthesis of β-amino acids are summarized. Then, the mechanisms of β- amino acid incorporation into natural products are reviewed. Because it is anticipated that the rational swapping of the β-amino acid moieties with various side chains and stereochemistries by biosynthetic engineering should lead to the creation of novel architectures and bioactive compounds, the accumulation of knowledge regarding β- amino acid-containing natural product biosynthetic machinery could have a significant impact in this field. -

Active Site Tyrosine Is Essential for Amidohydrolase but Not for Esterase
Active site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme Kristin Moreth, Daniel Riester, Christian Hildmann, René Hempel, Dennis Wegener, Andreas Schober, Andreas Schwienhorst To cite this version: Kristin Moreth, Daniel Riester, Christian Hildmann, René Hempel, Dennis Wegener, et al.. Ac- tive site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme. Biochemical Journal, Portland Press, 2006, 401 (3), pp.659-665. 10.1042/BJ20061239. hal-00478649 HAL Id: hal-00478649 https://hal.archives-ouvertes.fr/hal-00478649 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 12 Oct 2006 as manuscript BJ20061239 Active site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme Kristin Moreth¶, Daniel Riester¶, Christian Hildmann¶, René Hempel¶, Dennis Wegener¶,§,‡, -

Characterization of Aspartylglucosaminidase Activation and Aspartylglucosaminuria Mutations
Arto Pennanen Publications of the National Public Health Institute A 1 / 2004 — INDOOR AIR POLLUTION AND HEALTH RISKS IN FINNISH ICE ARENAS RISKSINFINNISHICE AND HEALTH AIR POLLUTION INDOOR Jani Saarela CHARACTERIZATION OF ASPARTYLGLUCOSAMINIDASE ACTIVATION AND ASPARTYLGLUCOSAMINURIA MUTATIONS ISBN 951-740-485-9 ISSN 0359-3584 ISBN 951-740-486-7 (pdf) Department of Molecular Medicine, ISSN 1458-6290 (pdf) National Public Health Institute, Helsinki, Finland and http://www.ktl.fi /portal/suomi/julkaisut/julkaisusarjat/ Department of Medical Genetics, kansanterveyslaitoksen_julkaisuja_a/ University of Helsinki, Finland Kopijyvä Kuopio 2005 Helsinki 2004 PPennanen_kansi.inddennanen_kansi.indd 1 117.2.20057.2.2005 115:26:195:26:19 CHARACTERIZATION OF ASPARTYLGLUCOSAMINIDASE ACTIVATION AND ASPARTYLGLUCOSAMINURIA MUTATIONS Jani Saarela Department of Molecular Medicine, National Public Health Institute, Helsinki, Finland and Department of Medical Genetics, University of Helsinki, Finland Academic Dissertation To be publicly discussed with the permission of the Medical Faculty of the University of Helsinki, in the lecture room 3 of Biomedicum Helsinki, Haartmaninkatu 8, Helsinki, on January 30th, 2004, at 12 o’clock noon. Helsinki 2004 Supervised by Professor Leena Peltonen-Palotie National Public Health Institute and Department of Medical Genetics University of Helsinki, Helsinki, Finland Reviewed by Professor Ole Kristian Tollersrud and Docent Marc Baumann Department of Medical Biochemistry Protein Chemistry/Proteomics Unit University of Tromsoe and Neuroscience Research Program Tromsoe, Norway University of Helsinki, Helsinki, Finland To be publicly discussed with Professor Marja Makarow Institute of Biotechnology and Department of Applied Biochemistry and Molecular Biology University of Helsinki, Helsinki, Finland Julkaisija-Utgivare-Publisher Kansanterveyslaitos (KTL) Mannerheimintie 166 00300 Helsinki puh. vaihde 09-47441, felefax 09-4744 8408 Folkhälsoinstitutet Mannerheimvägen 166 00300, Helsinki tel. -

Eira Kelo | Kelo | 156 | Eira
View metadata, citation and similar papers at core.ac.uk brought to you by CORE dissertations provided by UEF Electronic Publications | 156 | Eira| 156 Kelo | Eira Kelo Catalytic and Therapeutic Characteristics of Human Aspartylglycosaminuria (AGU), Recombinant Glycosylasparaginase an inherited lysosomal storage Catalytic and Therapeutic Characteristics Human of Recombinant Glycosylasparaginase and Bacterial L-asparaginases Eira Kelo disease, is caused by the deficient and Bacterial L-asparaginases activity of a lysosomal enzyme glycosylasparaginase (GA). Catalytic and Therapeutic Loss of GA activity results in the Characteristics of Human accumulation of glycoasparagines in tissues leading to progressive Recombinant Glycosylasparaginase psychomotor retardation and a shortened life span. Currently there is no cure for and Bacterial L-asparaginases AGU. In this study, the effectiveness of enzyme replacement therapy (ERT) with human recombinant GA was evaluated in the mouse model of AGU. Furthermore, the enzymatic properties of GA and bacterial asparaginases were compared. This study reveals new therapeutic and catalytic properties of human GA and bacterial asparaginases. Publications of the University of Eastern Finland Dissertations in Health Sciences Publications of the University of Eastern Finland Dissertations in Health Sciences isbn 978-952-61-1045-5 EIRA KELO EIRA KELO Catalytic and therapeutic characteristics of Catalytic and therapeutic characteristics of human recombinant glycosylasparaginase human recombinant glycosylasparaginase -

Chemistry of Acetyl Transfer by Histone Modifying Enzymes: Structure, Mechanism and Implications for Effector Design
Oncogene (2007) 26, 5528–5540 & 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00 www.nature.com/onc REVIEW Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design SC Hodawadekar and R Marmorstein The Wistar Institute and The Department of Chemistry, University of Pennsylvania, Philadelphia, PA, USA The post-translational modification of histones plays an remodeling proteins that mobilize the histone proteins important role in chromatin regulation, a process that within chromatin (Varga-Weisz and Becker, 2006); insures the fidelity of gene expression and other DNA histone chaperone proteins that assemble, disassemble transactions. Of the enzymes that mediate post-transla- or replace variant histones within chromatin (Loyola tion modification, the histone acetyltransferase (HAT) and Almouzni, 2004); and post-translational modifica- and histone deacetylase (HDAC) proteins that add and tion enzymes that add or remove functional groups to or remove acetyl groups to and from target lysine residues from the histone proteins (Nightingale et al., 2006). within histones, respectively, have been the most exten- The post-translational modifications of histones sively studied at both the functional and structural levels. involve the addition or removal of acetyl, methyl or Not surprisingly, the aberrant activity of several of these phosphate groups as well as the reversible transfer of the enzymes have been implicated in human diseases such as ubiquitin and sumo proteins. -

European Patent Office U.S. Patent and Trademark Office
EUROPEAN PATENT OFFICE U.S. PATENT AND TRADEMARK OFFICE CPC NOTICE OF CHANGES 89 DATE: JULY 1, 2015 PROJECT RP0098 The following classification changes will be effected by this Notice of Changes: Action Subclass Group(s) Symbols deleted: C12Y 101/01063 C12Y 101/01128 C12Y 101/01161 C12Y 102/0104 C12Y 102/03011 C12Y 103/01004 C12Y 103/0103 C12Y 103/01052 C12Y 103/99007 C12Y 103/9901 C12Y 103/99013 C12Y 103/99021 C12Y 105/99001 C12Y 105/99002 C12Y 113/11013 C12Y 113/12012 C12Y 114/15002 C12Y 114/99028 C12Y 204/01119 C12Y 402/01052 C12Y 402/01058 C12Y 402/0106 C12Y 402/01061 C12Y 601/01025 C12Y 603/02027 Symbols newly created: C12Y 101/01318 C12Y 101/01319 C12Y 101/0132 C12Y 101/01321 C12Y 101/01322 C12Y 101/01323 C12Y 101/01324 C12Y 101/01325 C12Y 101/01326 C12Y 101/01327 C12Y 101/01328 C12Y 101/01329 C12Y 101/0133 C12Y 101/01331 C12Y 101/01332 C12Y 101/01333 CPC Form – v.4 CPC NOTICE OF CHANGES 89 DATE: JULY 1, 2015 PROJECT RP0098 Action Subclass Group(s) C12Y 101/01334 C12Y 101/01335 C12Y 101/01336 C12Y 101/01337 C12Y 101/01338 C12Y 101/01339 C12Y 101/0134 C12Y 101/01341 C12Y 101/01342 C12Y 101/03043 C12Y 101/03044 C12Y 101/98003 C12Y 101/99038 C12Y 102/01083 C12Y 102/01084 C12Y 102/01085 C12Y 102/01086 C12Y 103/01092 C12Y 103/01093 C12Y 103/01094 C12Y 103/01095 C12Y 103/01096 C12Y 103/01097 C12Y 103/0701 C12Y 103/08003 C12Y 103/08004 C12Y 103/08005 C12Y 103/08006 C12Y 103/08007 C12Y 103/08008 C12Y 103/08009 C12Y 103/99032 C12Y 104/01023 C12Y 104/01024 C12Y 104/03024 C12Y 105/01043 C12Y 105/01044 C12Y 105/01045 C12Y 105/03019 C12Y 105/0302 -

Towards Combinatorial Biosynthesis of Pyrrolamide Antibiotics in Streptomyces Celine Aubry
Towards combinatorial biosynthesis of pyrrolamide antibiotics in Streptomyces Celine Aubry To cite this version: Celine Aubry. Towards combinatorial biosynthesis of pyrrolamide antibiotics in Streptomyces. Bio- chemistry [q-bio.BM]. Université Paris Saclay (COmUE), 2019. English. NNT : 2019SACLS245. tel-02955510 HAL Id: tel-02955510 https://tel.archives-ouvertes.fr/tel-02955510 Submitted on 2 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Towards combinatorial biosynthesis of pyrrolamide antibiotics in Streptomyces Thèse de doctorat de l'Université Paris-Saclay préparée à l’Université Paris-Sud École doctorale n°577 Structure et Dynamique des Systèmes Vivants (SDSV) Spécialité de doctorat : Sciences de la vie et de la Santé Thèse présentée et soutenue à Orsay, le 30/09/19, par Céline AUBRY Composition du Jury : Matthieu Jules Professeur, Agroparistech (MICALIS) Président du Jury Yanyan Li Chargée de recherche, MNHN (MCAM) Rapportrice Stéphane Cociancich Chercheur, CIRAD (BGPI) Rapporteur Annick Méjean Professeure, Université Paris-Diderot (LIED) Examinatrice Hasna Boubakri Maitre de conférences, Université Claude Bernard Lyon I (Ecologie microbienne) Examinatrice Sylvie Lautru Chargée de recherche, CNRS (I2BC) Directrice de thèse Acknowledgements J’ai insisté pour rédiger l’ensemble de ma thèse en anglais. -

Characterizing the Roles of Exit Tunnel Residues in Ligand Binding
CHARACTERIZING THE ROLES OF EXIT TUNNEL RESIDUES IN LIGAND BINDING AND CATALYSIS OF HISTONE DEACETYLASE-8 A Thesis Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Ruchi Gupta In Partial Fulfillment for the Degree of MASTER OF SCIENCE Major Department: Chemistry and Biochemistry March 2014 Fargo, North Dakota North Dakota State University Graduate School Title Characterizing the Roles of Exit Tunnel Residues in Ligand Binding and Catalysis of Histone Deacetylase-8 By Ruchi Gupta The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of MASTER OF SCIENCE SUPERVISORY COMMITTEE: Dr. D.K. Srivastava Chair Dr. Gregory Cook Dr. Stuart Haring Dr. Jane Schuh Approved: 03/26/2014 Gregory Cook Date Department Chair ABSTRACT Histone deacetylases are an important class of enzymes that catalyze the hydrolysis of acetyl-L-lysine side chains in histone and non-histone proteins to yield L-lysine and acetate, effecting the epigenetic regulation of gene expression. In addition to the active site pocket, the enzyme harbors an internal cavity for the release of acetate by-product. To probe the role of highly conserved amino acid residues lining this exit tunnel, site-directed alanine substitutions were made at tyrosine-18, tyrosine-20 and histidine-42 positions. These mutants were characterized by various biochemical and biophysical techniques to define the effect of mutations on ligand binding and catalysis of the enzyme. The mutations altered the catalytic activity of HDAC8 significantly. Y18A mutation dramatically impaired the structural-functional aspects of the enzymatic reaction. -

Hdacs, Histone Deacetylation and Gene Transcription: from Molecular Biology to Cancer Therapeutics
Paola Gallinari et al. npg Cell Research (2007) 17:195-211. npg195 © 2007 IBCB, SIBS, CAS All rights reserved 1001-0602/07 $ 30.00 REVIEW www.nature.com/cr HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics Paola Gallinari1, Stefania Di Marco1, Phillip Jones1, Michele Pallaoro1, Christian Steinkühler1 1Istituto di Ricerche di Biologia Molecolare “P. Angeletti”-IRBM-Merck Research Laboratories Rome, Via Pontina Km 30,600, 00040 Pomezia, Italy Histone deacetylases (HDACs) and histone acetyl transferases (HATs) are two counteracting enzyme families whose enzymatic activity controls the acetylation state of protein lysine residues, notably those contained in the N-terminal extensions of the core histones. Acetylation of histones affects gene expression through its influence on chromatin confor- mation. In addition, several non-histone proteins are regulated in their stability or biological function by the acetylation state of specific lysine residues. HDACs intervene in a multitude of biological processes and are part of a multiprotein family in which each member has its specialized functions. In addition, HDAC activity is tightly controlled through targeted recruitment, protein-protein interactions and post-translational modifications. Control of cell cycle progression, cell survival and differentiation are among the most important roles of these enzymes. Since these processes are affected by malignant transformation, HDAC inhibitors were developed as antineoplastic drugs and are showing encouraging efficacy in cancer patients. Keywords: histone deacetylase, histone, post-translational modification, transcription, histone deacetylase inhibitors, protein acetylation Cell Research (2007) 17:195-211. doi: 10.1038/sj.cr.7310149; published online 27 February 2007 Introduction tone deacetylase (HDAC) family of chromatin-modifying enzymes.