Characterization of Aspartylglucosaminidase Activation and Aspartylglucosaminuria Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

Characterization of Α-L-Fucosidase and Other Digestive Hydrolases From
Acta Tropica 141 (2015) 118–127 Contents lists available at ScienceDirect Acta Tropica journal homepage: www.elsevier.com/locate/actatropica Characterization of ␣-L-fucosidase and other digestive hydrolases from Biomphalaria glabrata Natalia N. Perrella a,b, Rebeca S. Cantinha c,d, Eliana Nakano c, Adriana R. Lopes a,∗ a Laboratory of Biochemistry and Biophysics—Instituto Butantan, São Paulo, Brazil b Programa de Pós Graduac¸ ão Interunidades em Biotecnologia PPIB, Universidade de São Paulo, São Paulo, SP, Brazil c Laboratory of Parasitology—Instituto Butantan, São Paulo, Brazil d Instituto de Pesquisas Energéticas e Nucleares, Universidade de São Paulo, São Paulo, SP, Brazil article info abstract Article history: Schistosoma mansoni is one of the major agents of the disease Schistosomiasis, which is one of the Received 10 February 2014 major global public health concerns. Biomphalaria glabrata is an obligate intermediate mollusc host of Received in revised form 3 July 2014 S. mansoni. Although the development of S. mansoni occurs in the snail hepatopancreas, studies that Accepted 12 August 2014 focus on this organ remain limited. In this study, we biochemically identified five distinct carbohy- Available online 16 September 2014 drases (amylase, maltase, ␣-glucosidase, trehalase, and ␣-L-fucosidase), lipases, and peptidases in the B. glabrata hepatopancreas and focused on the isolation and characterization of the activity of ␣-L- Keywords: fucosidase. The isolated ␣-L-fucosidase has a molecular mass of 141 kDa, an optimum pH of 5.8, and Hepatopancreas ␣ Enzymes is inhibited by Tris, fucose, and 1-deoxyfuconojirimycin. B. glabrata -L-fucosidase is an exoglycosidase ␣-L-Fucosidase that can hydrolyze the natural substrate fucoidan to fucose residues. -

Glycoproteomics-Based Signatures for Tumor Subtyping and Clinical Outcome Prediction of High-Grade Serous Ovarian Cancer
ARTICLE https://doi.org/10.1038/s41467-020-19976-3 OPEN Glycoproteomics-based signatures for tumor subtyping and clinical outcome prediction of high-grade serous ovarian cancer Jianbo Pan 1,2,3, Yingwei Hu1,3, Shisheng Sun 1,3, Lijun Chen1, Michael Schnaubelt1, David Clark1, ✉ Minghui Ao1, Zhen Zhang1, Daniel Chan1, Jiang Qian2 & Hui Zhang 1 1234567890():,; Inter-tumor heterogeneity is a result of genomic, transcriptional, translational, and post- translational molecular features. To investigate the roles of protein glycosylation in the heterogeneity of high-grade serous ovarian carcinoma (HGSC), we perform mass spectrometry-based glycoproteomic characterization of 119 TCGA HGSC tissues. Cluster analysis of intact glycoproteomic profiles delineates 3 major tumor clusters and 5 groups of intact glycopeptides. It also shows a strong relationship between N-glycan structures and tumor molecular subtypes, one example of which being the association of fucosylation with mesenchymal subtype. Further survival analysis reveals that intact glycopeptide signatures of mesenchymal subtype are associated with a poor clinical outcome of HGSC. In addition, we study the expression of mRNAs, proteins, glycosites, and intact glycopeptides, as well as the expression levels of glycosylation enzymes involved in glycoprotein biosynthesis pathways in each tumor. The results show that glycoprotein levels are mainly controlled by the expression of their individual proteins, and, furthermore, that the glycoprotein-modifying glycans cor- respond to the protein levels of glycosylation enzymes. The variation in glycan types further shows coordination to the tumor heterogeneity. Deeper understanding of the glycosylation process and glycosylation production in different subtypes of HGSC may provide important clues for precision medicine and tumor-targeted therapy. -
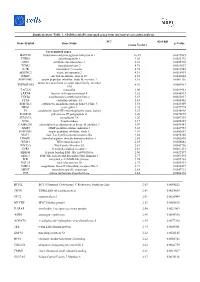
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Biochemical Characterization and Comparison of Aspartylglucosaminidases Secreted in Venom of the Parasitoid Wasps Asobara Tabida and Leptopilina Heterotoma
RESEARCH ARTICLE Biochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma Quentin Coulette1, SeÂverine Lemauf2, Dominique Colinet2, Geneviève PreÂvost1, Caroline Anselme1, Marylène Poirie 2, Jean-Luc Gatti2* a1111111111 1 Unite ªEcologie et Dynamique des Systèmes AnthropiseÂsº (EDYSAN, FRE 3498 CNRS-UPJV), Universite de Picardie Jules Verne, Amiens, France, 2 Universite CoÃte d'Azur, INRA, CNRS, ISA, Sophia Antipolis, a1111111111 France a1111111111 a1111111111 * [email protected] a1111111111 Abstract Aspartylglucosaminidase (AGA) is a low-abundance intracellular enzyme that plays a key OPEN ACCESS role in the last stage of glycoproteins degradation, and whose deficiency leads to human Citation: Coulette Q, Lemauf S, Colinet D, PreÂvost aspartylglucosaminuria, a lysosomal storage disease. Surprisingly, high amounts of AGA- G, Anselme C, Poirie M, et al. (2017) Biochemical characterization and comparison of like proteins are secreted in the venom of two phylogenetically distant hymenopteran para- aspartylglucosaminidases secreted in venom of the sitoid wasp species, Asobara tabida (Braconidae) and Leptopilina heterotoma (Cynipidae). parasitoid wasps Asobara tabida and Leptopilina These venom AGAs have a similar domain organization as mammalian AGAs. They share heterotoma. PLoS ONE 12(7): e0181940. https:// with them key residues for autocatalysis and activity, and the mature - and -subunits also doi.org/10.1371/journal.pone.0181940 α β form an (αβ)2 structure in solution. Interestingly, only one of these AGAs subunits (α for Editor: Erjun Ling, Institute of Plant Physiology and AtAGA and for LhAGA) is glycosylated instead of the two subunits for lysosomal human Ecology Shanghai Institutes for Biological β Sciences, CHINA AGA (hAGA), and these glycosylations are partially resistant to PGNase F treatment. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Biosynthesis of Natural Products Containing Β-Amino Acids
Natural Product Reports Biosynthesis of natural products containing β -amino acids Journal: Natural Product Reports Manuscript ID: NP-REV-01-2014-000007.R1 Article Type: Review Article Date Submitted by the Author: 21-Apr-2014 Complete List of Authors: Kudo, Fumitaka; Tokyo Institute Of Technology, Department of Chemistry Miyanaga, Akimasa; Tokyo Institute Of Technology, Department of Chemistry Eguchi, T; Tokyo Institute Of Technology, Department of Chemistry and Materials Science Page 1 of 20 Natural Product Reports NPR RSC Publishing REVIEW Biosynthesis of natural products containing βββ- amino acids Cite this: DOI: 10.1039/x0xx00000x Fumitaka Kudo, a Akimasa Miyanaga, a and Tadashi Eguchi *b Received 00th January 2014, We focus here on β-amino acids as components of complex natural products because the presence of β-amino acids Accepted 00th January 2014 produces structural diversity in natural products and provides characteristic architectures beyond that of ordinary DOI: 10.1039/x0xx00000x α-L-amino acids, thus generating significant and unique biological functions in nature. In this review, we first survey the known bioactive β-amino acid-containing natural products including nonribosomal peptides, www.rsc.org/ macrolactam polyketides, and nucleoside-β-amino acid hybrids. Next, the biosynthetic enzymes that form β-amino acids from α-amino acids and de novo synthesis of β-amino acids are summarized. Then, the mechanisms of β- amino acid incorporation into natural products are reviewed. Because it is anticipated that the rational swapping of the β-amino acid moieties with various side chains and stereochemistries by biosynthetic engineering should lead to the creation of novel architectures and bioactive compounds, the accumulation of knowledge regarding β- amino acid-containing natural product biosynthetic machinery could have a significant impact in this field. -

Common Differentially Expressed Genes and Pathways Correlating Both Coronary Artery Disease and Atrial Fibrillation
EXCLI Journal 2021;20:126-141– ISSN 1611-2156 Received: December 08, 2020, accepted: January 11, 2021, published: January 18, 2021 Supplementary material to: Original article: COMMON DIFFERENTIALLY EXPRESSED GENES AND PATHWAYS CORRELATING BOTH CORONARY ARTERY DISEASE AND ATRIAL FIBRILLATION Youjing Zheng, Jia-Qiang He* Department of Biomedical Sciences and Pathobiology, College of Veterinary Medicine, Virginia Tech, Blacksburg, VA 24061, USA * Corresponding author: Jia-Qiang He, Department of Biomedical Sciences and Pathobiology, Virginia Tech, Phase II, Room 252B, Blacksburg, VA 24061, USA. Tel: 1-540-231-2032. E-mail: [email protected] https://orcid.org/0000-0002-4825-7046 Youjing Zheng https://orcid.org/0000-0002-0640-5960 Jia-Qiang He http://dx.doi.org/10.17179/excli2020-3262 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/). Supplemental Table 1: Abbreviations used in the paper Abbreviation Full name ABCA5 ATP binding cassette subfamily A member 5 ABCB6 ATP binding cassette subfamily B member 6 (Langereis blood group) ABCB9 ATP binding cassette subfamily B member 9 ABCC10 ATP binding cassette subfamily C member 10 ABCC13 ATP binding cassette subfamily C member 13 (pseudogene) ABCC5 ATP binding cassette subfamily C member 5 ABCD3 ATP binding cassette subfamily D member 3 ABCE1 ATP binding cassette subfamily E member 1 ABCG1 ATP binding cassette subfamily G member 1 ABCG4 ATP binding cassette subfamily G member 4 ABHD18 Abhydrolase domain -

Keyword Index
Gene Therapy (1998) 5, 1717–1719 1998 Stockton Press All rights reserved 0969-7128/98 $12.00 http://www.stockton-press.co.uk/gt Keyword Index Volume 5 1998 15-deoxyspergualin 85 (BC) bcl-2 1156 complex stability 137 (BC) 293 cells adenoviral vectors 563 (BC) bicistronic vectors 1566 (BC) computerized tomography 896 5-azacytidine 1299 bidirectional promoter 76 connexin43 1114, 1221, 1372 5-fluorocytosine 507 biodegradable microspheres 740 cord blood 233 6-hydroxydopamine (6-OHDA) 1650 biolistics 692 corneal endothelium 1347 99mTc 798 BMP-7 1098 coronary infusion 1472 bone repair 1098 costimulation 223 AAV 50, 820 bradykinin 630 costimulatory 965 AAV helper genes 938 brain 712 (BC), 820 Cre-mediated recombination 1047 AAV vector production 938 brain tumors 1122 Cre/loxP 684 adeno-associated virus 277 (BC), 1434, breast tumor 240 cyclophosphamide 189 1604, 1642 bronchoscopy 166 cystic fibrosis 91, 131 (BC), 181, 218, 309, adeno-associated virus (AAV) 166 bystander effect 105, 113, 605, 1114, 1221, 345, 583, 1488 adenosine deaminase 320 1372, 1705 cytidine deaminase 1545 adenoviral gene transfer 85 cytochrome P450 1070 adenoviral vectors 309, 869, 1251 C2C12 1014 cytokine gene 1462 adenovirus 8, 19, 131 (BC), 174, 339, 345, CAG 240 cytokine gene transfer 481 352, 473, 507, 605, 621, 630, 740, 747, cancer 728 (R), 1105, 1400, 1462 cytokine receptors 253 778, 938, 975, 995, 1023, 1038, 1130, cancer gene therapy 189, 451, 1171 cytokines 253, 869, 1400 1156, 1213, 1259, 1314, 1363, 1400, 1552, cancer vaccine 247, 1677 cytosine arabinoside 1545 -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in