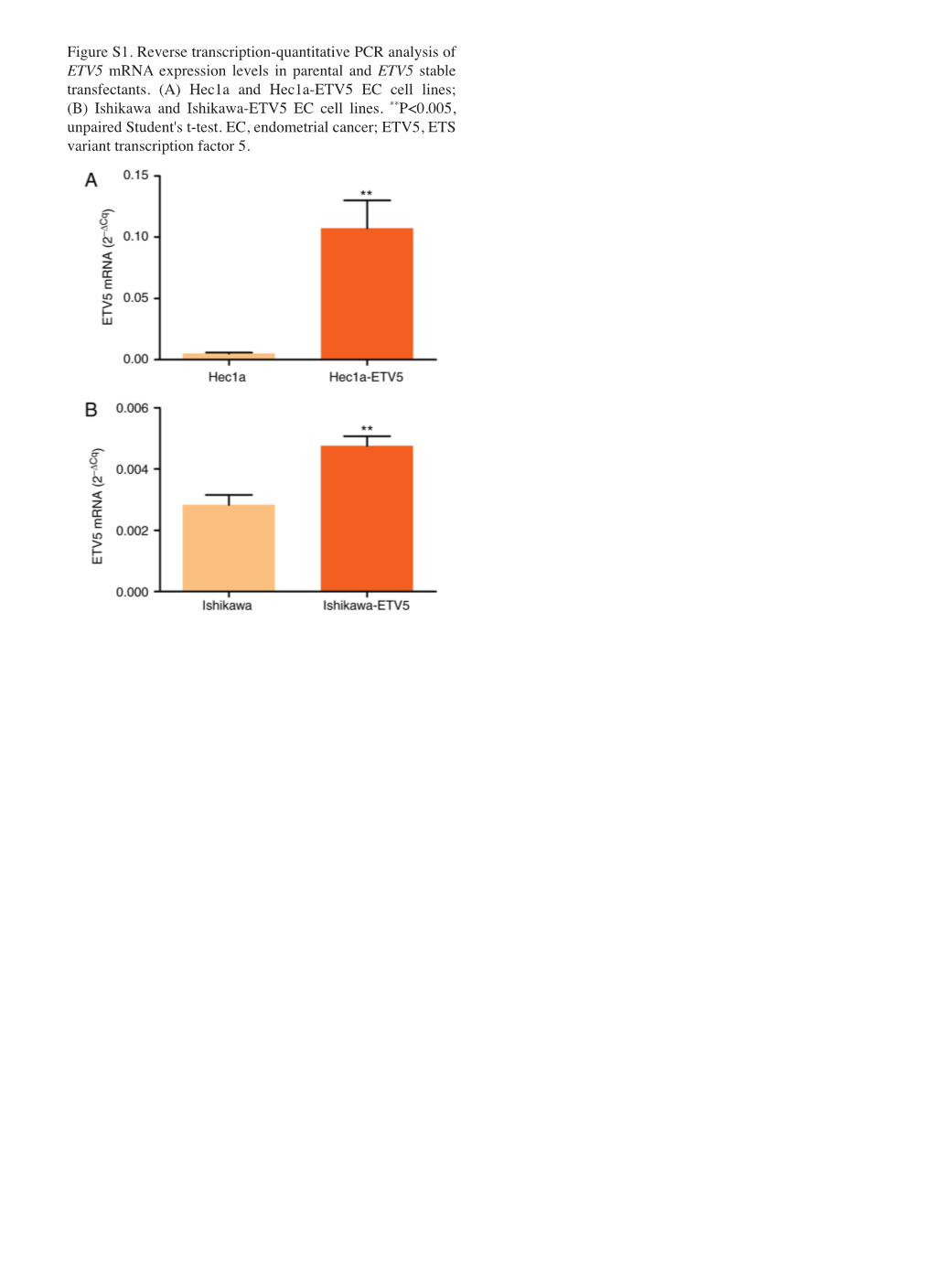
Figure S1. Reverse Transcription‑Quantitative PCR Analysis of ETV5 Mrna Expression Levels in Parental and ETV5 Stable Transfectants
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aberrant Methylation Underlies Insulin Gene Expression in Human Insulinoma
ARTICLE https://doi.org/10.1038/s41467-020-18839-1 OPEN Aberrant methylation underlies insulin gene expression in human insulinoma Esra Karakose1,6, Huan Wang 2,6, William Inabnet1, Rajesh V. Thakker 3, Steven Libutti4, Gustavo Fernandez-Ranvier 1, Hyunsuk Suh1, Mark Stevenson 3, Yayoi Kinoshita1, Michael Donovan1, Yevgeniy Antipin1,2, Yan Li5, Xiaoxiao Liu 5, Fulai Jin 5, Peng Wang 1, Andrew Uzilov 1,2, ✉ Carmen Argmann 1, Eric E. Schadt 1,2, Andrew F. Stewart 1,7 , Donald K. Scott 1,7 & Luca Lambertini 1,6 1234567890():,; Human insulinomas are rare, benign, slowly proliferating, insulin-producing beta cell tumors that provide a molecular “recipe” or “roadmap” for pathways that control human beta cell regeneration. An earlier study revealed abnormal methylation in the imprinted p15.5-p15.4 region of chromosome 11, known to be abnormally methylated in another disorder of expanded beta cell mass and function: the focal variant of congenital hyperinsulinism. Here, we compare deep DNA methylome sequencing on 19 human insulinomas, and five sets of normal beta cells. We find a remarkably consistent, abnormal methylation pattern in insu- linomas. The findings suggest that abnormal insulin (INS) promoter methylation and altered transcription factor expression create alternative drivers of INS expression, replacing cano- nical PDX1-driven beta cell specification with a pathological, looping, distal enhancer-based form of transcriptional regulation. Finally, NFaT transcription factors, rather than the cano- nical PDX1 enhancer complex, are predicted to drive INS transactivation. 1 From the Diabetes Obesity and Metabolism Institute, The Department of Surgery, The Department of Pathology, The Department of Genetics and Genomics Sciences and The Institute for Genomics and Multiscale Biology, The Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA. -

Aurora Kinase a in Gastrointestinal Cancers: Time to Target Ahmed Katsha1, Abbes Belkhiri1, Laura Goff3 and Wael El-Rifai1,2,4*
Katsha et al. Molecular Cancer (2015) 14:106 DOI 10.1186/s12943-015-0375-4 REVIEW Open Access Aurora kinase A in gastrointestinal cancers: time to target Ahmed Katsha1, Abbes Belkhiri1, Laura Goff3 and Wael El-Rifai1,2,4* Abstract Gastrointestinal (GI) cancers are a major cause of cancer-related deaths. During the last two decades, several studies have shown amplification and overexpression of Aurora kinase A (AURKA) in several GI malignancies. These studies demonstrated that AURKA not only plays a role in regulating cell cycle and mitosis, but also regulates a number of key oncogenic signaling pathways. Although AURKA inhibitors have moved to phase III clinical trials in lymphomas, there has been slower progress in GI cancers and solid tumors. Ongoing clinical trials testing AURKA inhibitors as a single agent or in combination with conventional chemotherapies are expected to provide important clinical information for targeting AURKA in GI cancers. It is, therefore, imperative to consider investigations of molecular determinants of response and resistance to this class of inhibitors. This will improve evaluation of the efficacy of these drugs and establish biomarker based strategies for enrollment into clinical trials, which hold the future direction for personalized cancer therapy. In this review, we will discuss the available data on AURKA in GI cancers. We will also summarize the major AURKA inhibitors that have been developed and tested in pre-clinical and clinical settings. Keywords: Aurora kinases, Therapy, AURKA inhibitors, MNL8237, Alisertib, Gastrointestinal, Cancer, Signaling pathways Introduction stage [9-11]. Furthermore, AURKA is critical for Mitotic kinases are the main proteins that coordinate ac- bipolar-spindle assembly where it interacts with Ran- curate mitotic processing [1]. -
Nucleoporin 107, 62 and 153 Mediate Kcnq1ot1 Imprinted Domain Regulation in Extraembryonic Endoderm Stem Cells
ARTICLE DOI: 10.1038/s41467-018-05208-2 OPEN Nucleoporin 107, 62 and 153 mediate Kcnq1ot1 imprinted domain regulation in extraembryonic endoderm stem cells Saqib S. Sachani 1,2,3,4, Lauren S. Landschoot1,2, Liyue Zhang1,2, Carlee R. White1,2, William A. MacDonald3,4, Michael C. Golding 5 & Mellissa R.W. Mann 3,4 1234567890():,; Genomic imprinting is a phenomenon that restricts transcription to predominantly one par- ental allele. How this transcriptional duality is regulated is poorly understood. Here we perform an RNA interference screen for epigenetic factors involved in paternal allelic silen- cing at the Kcnq1ot1 imprinted domain in mouse extraembryonic endoderm stem cells. Multiple factors are identified, including nucleoporin 107 (NUP107). To determine NUP107’s role and specificity in Kcnq1ot1 imprinted domain regulation, we deplete Nup107, as well as Nup62, Nup98/96 and Nup153. Nup107, Nup62 and Nup153, but not Nup98/96 depletion, reduce Kcnq1ot1 noncoding RNA volume, displace the Kcnq1ot1 domain from the nuclear periphery, reactivate a subset of normally silent paternal alleles in the domain, alter histone modifications with concomitant changes in KMT2A, EZH2 and EHMT2 occupancy, as well as reduce cohesin interactions at the Kcnq1ot1 imprinting control region. Our results establish an important role for specific nucleoporins in mediating Kcnq1ot1 imprinted domain regulation. 1 Departments of Obstetrics & Gynaecology, and Biochemistry, Western University, Schulich School of Medicine and Dentistry, London, ON N6A 5W9, Canada. 2 Children’s Health Research Institute, London, ON N6C 2V5, Canada. 3 Departments of Obstetrics, Gynecology and Reproductive Sciences, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA. 4 Magee-Womens Research Institute, Pittsburgh, PA 15213, USA. -

Basic Helix-Loop-Helix Transcription Factors DEC1 and DEC2 Regulate the Paclitaxel-Induced Apoptotic Pathway of MCF-7 Human Breast Cancer Cells
INTERNATIONAL JOURNAL OF MoleCular MEDICine 27: 491-495, 2011 Basic helix-loop-helix transcription factors DEC1 and DEC2 regulate the paclitaxel-induced apoptotic pathway of MCF-7 human breast cancer cells YUNYAN WU1,2, FUYUKI SATO1, UJJAL KUMAR BHAWAL3, TAKESHI KAWAMOTO4, KATSUMI FUJIMOTO4, MITSUHIDE NOSHIRO4, SATOKO MOROHASHI1, YUKIO KATO4 and HIROSHI KIJIMA1 1Department of Pathology and Bioscience, Hirosaki University Graduate School of Medicine, Hirosaki 036-8562, Japan; 2Department of Pathology, College of Basic Medical Sciences, China Medical University, Shenyang 110001, P.R. China; 3Department of Oral and Maxillofacial Surgery and High-Tech Research Center, Kanagawa Dental College, Yokosuka 238-8580; 4Department of Dental and Medical Biochemistry, Hiroshima University Graduate School of Biomedical Science, Hiroshima 734-8553, Japan Received November 17, 2010; Accepted January 10, 2011 DOI: 10.3892/ijmm.2011.617 Abstract. Differentiated embryonic chondrocyte gene Introduction (DEC) 1 (BHLHE40/Stra13/Sharp2) and DEC2 (BHLHE41/ Sharp1) are basic helix-loop-helix (bHLH) transcription Paclitaxel is an anti-tumor drug that is used against a wide factors that are associated with the regulation of apoptosis, cell variety of solid tumors (1,2) and affects the expression of proliferation and circadian rhythms, as well as malignancy Bcl-2, Bcl-xL and phosphorylated-c-Jun NH2-terminal kinase in various cancers. However, the roles of DEC1 and DEC2 (JNK), and the activation of caspases and poly (ADP-ribose) expression in breast cancer are poorly understood. In this polymerase PARP (3-6), resulting in the induction of apop- study, we sought to examine the roles of DEC1 and DEC2 tosis by p53-dependent or -independent mechanisms (7-9). -

RASSF1A Interacts with and Activates the Mitotic Kinase Aurora-A
Oncogene (2008) 27, 6175–6186 & 2008 Macmillan Publishers Limited All rights reserved 0950-9232/08 $32.00 www.nature.com/onc ORIGINAL ARTICLE RASSF1A interacts with and activates the mitotic kinase Aurora-A L Liu1, C Guo1, R Dammann2, S Tommasi1 and GP Pfeifer1 1Division of Biology, Beckman Research Institute, City of Hope Cancer Center, Duarte, CA, USA and 2Institute of Genetics, University of Giessen, Giessen, Germany The RAS association domain family 1A (RASSF1A) gene tumorigenesis and carcinogen-induced tumorigenesis is located at chromosome 3p21.3 within a specific area of (Tommasi et al., 2005; van der Weyden et al., 2005), common heterozygous and homozygous deletions. RASS- supporting the notion that RASSF1A is a bona fide F1A frequently undergoes promoter methylation-asso- tumor suppressor. However, it is not fully understood ciated inactivation in human cancers. Rassf1aÀ/À mice how RASSF1A is involved in tumor suppression. are prone to both spontaneous and carcinogen-induced The biochemical function of the RASSF1A protein is tumorigenesis, supporting the notion that RASSF1A is a largely unknown. The homology of RASSF1A with the tumor suppressor. However, it is not fully understood how mammalian Ras effector novel Ras effector (NORE)1 RASSF1A is involved in tumor suppression pathways. suggests that the RASSF1A gene product may function Here we show that overexpression of RASSF1A inhibits in signal transduction pathways involving Ras-like centrosome separation. RASSF1A interacts with Aurora-A, proteins. However, recent data indicate that RASSF1A a mitotic kinase. Surprisingly, knockdown of RASS- itself binds to RAS only weakly and that binding to F1A by siRNA led to reduced activation of Aurora-A, RAS may require heterodimerization of RASSF1A and whereas overexpression of RASSF1A resulted in in- NORE1 (Ortiz-Vega et al., 2002). -

Downregulation of Dipeptidyl Peptidase 4 Accelerates Progression to Castration-Resistant Prostate Cancer Joshua W
Published OnlineFirst September 21, 2018; DOI: 10.1158/0008-5472.CAN-18-0687 Cancer Priority Report Research Downregulation of Dipeptidyl Peptidase 4 Accelerates Progression to Castration-Resistant Prostate Cancer Joshua W. Russo1,CeGao2, Swati S. Bhasin2, Olga S. Voznesensky1, Carla Calagua3, Seiji Arai1,4, Peter S. Nelson5, Bruce Montgomery6, Elahe A. Mostaghel5, Eva Corey6, Mary-Ellen Taplin7, Huihui Ye3, Manoj Bhasin2, and Steven P. Balk1 Abstract The standard treatment for metastatic prostate cancer, cleaves dipeptides from multiple growth factors, resulting in androgen deprivation therapy (ADT), is designed to suppress their increased degradation. DPP4 mRNA and protein were androgen receptor (AR) activity. However, men invariably also decreased in clinical CRPC cases, and inhibition of progress to castration-resistant prostate cancer (CRPC), and DPP4 with sitagliptin enhanced the growth of prostate AR reactivation contributes to progression in most cases. To cancer xenografts following castration. Significantly, DPP4 identify mechanisms that may drive CRPC, we examined a inhibitors are frequently used to treat type 2 diabetes as they VCaP prostate cancer xenograft model as tumors progressed increase insulin secretion. Together, these results implicate from initial androgen sensitivity prior to castration to castra- DPP4 as an AR-regulated tumor suppressor gene whose tion resistance and then on to relapse after combined therapy loss enhances growth factor activity and suggest that treat- with further AR-targeted drugs (abiraterone plus enzaluta- ment with DPP4 inhibitors may accelerate emergence of mide). AR activity persisted in castration-resistant and abir- resistance to ADT. aterone/enzalutamide–resistant xenografts and was associated with increased expression of the AR gene and the AR-V7 splice Significance: These findings identify DPP4 as an AR-stim- variant. -

Beta-Arrestin-Mediated Signaling in the Heart
SPECIAL ARTICLE Circ J 2008; 72: 1725–1729 Beta-Arrestin-Mediated Signaling in the Heart Priyesh A. Patel, BS; Douglas G. Tilley, PhD*; Howard A. Rockman, MD*,** Beta-arrestin is a multifunctional adapter protein well known for its role in G-protein-coupled receptor (GPCR) desensitization. Exciting new evidence indicates thatβ-arrestin is also a signaling molecule capable of initiating its own G-protein-independent signaling at GPCRs. One of the best-studiedβ-arrestin signaling pathways is the one involvingβ-arrestin-dependent activation of a mitogen-activated protein kinase cascade, the extracellular regulated kinase (ERK). ERK signaling, which is classically activated by agonist stimulation of the epidermal growth factor receptor (EGFR), can be activated by a number of GPCRs in aβ-arrestin-dependent manner. Recent work in animal models of heart failure suggests thatβ-arrestin-dependent activation of EGFR/ERK signaling by theβ-1-adrenergic receptor, and possibly the angiotensin II Type 1A receptor, are cardioprotective. Hence, a new model of signaling at cardiac GPCRs has emerged and implicates classical G-protein-mediated signaling with promoting harmful remodeling in heart failure, while concurrently linkingβ-arrestin-dependent, G-protein-inde- pendent signaling with cardioprotective effects. Based on this paradigm, a new class of drugs could be identified, termed “biased ligands”, which simultaneously block harmful G-protein signaling, while also promoting cardio- protectiveβ-arrestin-dependent signaling, leading to a potential breakthrough -

BMC Evolutionary Biology Biomed Central
BMC Evolutionary Biology BioMed Central Research article Open Access On the origins of arrestin and rhodopsin Carlos E Alvarez1,2,3 Address: 1Center for Molecular and Human Genetics, The Research Institute at Nationwide Children's Hospital, Columbus, OH, 43205, USA, 2Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH, 43210, USA and 3Novartis Institutes of BioMedical Research, CH-4002 Basel, Switzerland Email: Carlos E Alvarez - [email protected] Published: 29 July 2008 Received: 11 January 2008 Accepted: 29 July 2008 BMC Evolutionary Biology 2008, 8:222 doi:10.1186/1471-2148-8-222 This article is available from: http://www.biomedcentral.com/1471-2148/8/222 © 2008 Alvarez; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: G protein coupled receptors (GPCRs) are the most numerous proteins in mammalian genomes, and the most common targets of clinical drugs. However, their evolution remains enigmatic. GPCRs are intimately associated with trimeric G proteins, G protein receptor kinases, and arrestins. We conducted phylogenetic studies to reconstruct the history of arrestins. Those findings, in turn, led us to investigate the origin of the photosensory GPCR rhodopsin. Results: We found that the arrestin clan is comprised of the Spo0M protein family in archaea and bacteria, and the arrestin and Vps26 families in eukaryotes. The previously known animal arrestins are members of the visual/beta subfamily, which branched from the founding "alpha" arrestins relatively recently. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Automethylation of PRC2 Promotes H3K27 Methylation and Is Impaired in H3K27M Pediatric Glioma
Downloaded from genesdev.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma Chul-Hwan Lee,1,2,7 Jia-Ray Yu,1,2,7 Jeffrey Granat,1,2,7 Ricardo Saldaña-Meyer,1,2 Joshua Andrade,3 Gary LeRoy,1,2 Ying Jin,4 Peder Lund,5 James M. Stafford,1,2,6 Benjamin A. Garcia,5 Beatrix Ueberheide,3 and Danny Reinberg1,2 1Department of Biochemistry and Molecular Pharmacology, New York University School of Medicine, New York, New York 10016, USA; 2Howard Hughes Medical Institute, Chevy Chase, Maryland 20815, USA; 3Proteomics Laboratory, New York University School of Medicine, New York, New York 10016, USA; 4Shared Bioinformatics Core, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA; 5Department of Biochemistry and Molecular Biophysics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA The histone methyltransferase activity of PRC2 is central to the formation of H3K27me3-decorated facultative heterochromatin and gene silencing. In addition, PRC2 has been shown to automethylate its core subunits, EZH1/ EZH2 and SUZ12. Here, we identify the lysine residues at which EZH1/EZH2 are automethylated with EZH2-K510 and EZH2-K514 being the major such sites in vivo. Automethylated EZH2/PRC2 exhibits a higher level of histone methyltransferase activity and is required for attaining proper cellular levels of H3K27me3. While occurring inde- pendently of PRC2 recruitment to chromatin, automethylation promotes PRC2 accessibility to the histone H3 tail. Intriguingly, EZH2 automethylation is significantly reduced in diffuse intrinsic pontine glioma (DIPG) cells that carry a lysine-to-methionine substitution in histone H3 (H3K27M), but not in cells that carry either EZH2 or EED mutants that abrogate PRC2 allosteric activation, indicating that H3K27M impairs the intrinsic activity of PRC2. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US).