Chemistry of Acetyl Transfer by Histone Modifying Enzymes: Structure, Mechanism and Implications for Effector Design
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

Gentaur Products List
Chapter 2 : Gentaur Products List • Rabbit Anti LAMR1 Polyclonal Antibody Cy5 Conjugated Conjugated • Rabbit Anti Podoplanin gp36 Polyclonal Antibody Cy5 • Rabbit Anti LAMR1 CT Polyclonal Antibody Cy5 • Rabbit Anti phospho NFKB p65 Ser536 Polyclonal Conjugated Conjugated Antibody Cy5 Conjugated • Rabbit Anti CHRNA7 Polyclonal Antibody Cy5 Conjugated • Rat Anti IAA Monoclonal Antibody Cy5 Conjugated • Rabbit Anti EV71 VP1 CT Polyclonal Antibody Cy5 • Rabbit Anti Connexin 40 Polyclonal Antibody Cy5 • Rabbit Anti IAA Indole 3 Acetic Acid Polyclonal Antibody Conjugated Conjugated Cy5 Conjugated • Rabbit Anti LHR CGR Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Integrin beta 7 Polyclonal Antibody Cy5 • Rabbit Anti Natrexone Polyclonal Antibody Cy5 Conjugated • Rabbit Anti MMP 20 Polyclonal Antibody Cy5 Conjugated Conjugated • Rabbit Anti Melamine Polyclonal Antibody Cy5 Conjugated • Rabbit Anti BCHE NT Polyclonal Antibody Cy5 Conjugated • Rabbit Anti NAP1 NAP1L1 Polyclonal Antibody Cy5 • Rabbit Anti Acetyl p53 K382 Polyclonal Antibody Cy5 • Rabbit Anti BCHE CT Polyclonal Antibody Cy5 Conjugated Conjugated Conjugated • Rabbit Anti HPV16 E6 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti CCP Polyclonal Antibody Cy5 Conjugated • Rabbit Anti JAK2 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti HPV18 E6 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti HDC Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Microsporidia protien Polyclonal Antibody Cy5 • Rabbit Anti HPV16 E7 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Neurocan Polyclonal -

Interplay Between Epigenetics and Metabolism in Oncogenesis: Mechanisms and Therapeutic Approaches
OPEN Oncogene (2017) 36, 3359–3374 www.nature.com/onc REVIEW Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches CC Wong1, Y Qian2,3 and J Yu1 Epigenetic and metabolic alterations in cancer cells are highly intertwined. Oncogene-driven metabolic rewiring modifies the epigenetic landscape via modulating the activities of DNA and histone modification enzymes at the metabolite level. Conversely, epigenetic mechanisms regulate the expression of metabolic genes, thereby altering the metabolome. Epigenetic-metabolomic interplay has a critical role in tumourigenesis by coordinately sustaining cell proliferation, metastasis and pluripotency. Understanding the link between epigenetics and metabolism could unravel novel molecular targets, whose intervention may lead to improvements in cancer treatment. In this review, we summarized the recent discoveries linking epigenetics and metabolism and their underlying roles in tumorigenesis; and highlighted the promising molecular targets, with an update on the development of small molecule or biologic inhibitors against these abnormalities in cancer. Oncogene (2017) 36, 3359–3374; doi:10.1038/onc.2016.485; published online 16 January 2017 INTRODUCTION metabolic genes have also been identified as driver genes It has been appreciated since the early days of cancer research mutated in some cancers, such as isocitrate dehydrogenase 1 16 17 that the metabolic profiles of tumor cells differ significantly from and 2 (IDH1/2) in gliomas and acute myeloid leukemia (AML), 18 normal cells. Cancer cells have high metabolic demands and they succinate dehydrogenase (SDH) in paragangliomas and fuma- utilize nutrients with an altered metabolic program to support rate hydratase (FH) in hereditary leiomyomatosis and renal cell 19 their high proliferative rates and adapt to the hostile tumor cancer (HLRCC). -

Multiple N-Acetyltransferases and Drug Metabolism TISSUE DISTRIBUTION, CHARACTERIZATION and SIGNIFICANCE of MAMMALIAN N-ACETYLTRANSFERASE by D
Biochem. J. (1973) 132, 519-526 519 Printed in Great Britain Multiple N-Acetyltransferases and Drug Metabolism TISSUE DISTRIBUTION, CHARACTERIZATION AND SIGNIFICANCE OF MAMMALIAN N-ACETYLTRANSFERASE By D. J. HEARSE* and W. W. WEBER Department ofPharmacology, New York University Medical Center, 550 First Avenue, New York, N. Y. 10016, U.S.A. (Received 6 October 1972) Investigations in the rabbit have indicated the existence of more than one N-acetyl- transferase (EC 2.3.1.5). At least two enzymes, possibly isoenzymes, were partially characterized. The enzymes differed in their tissue distribution, substrate specificity, stability and pH characteristics. One of the enzymes was primarily associated with liver and gut and catalysed the acetylation of a wide range of drugs and foreign compounds, e.g. isoniazid, p-aminobenzoic acid, sulphamethazine and sulphadiazine. The activity of this enzyme corresponded to the well-characterized polymorphic trait of isoniazid acetylation, and determined whether individuals were classified as either 'rapid' or 'slow' acetylators. Another enzyme activity found in extrahepatic tissues readily catalysed the acetylation ofp-aminobenzoic acid but was much less active towards isoniazid and sulpha- methazine. The activity of this enzyme remained relatively constant from individual to individual. Studies in vitro and in vivo with both 'rapid' and 'slow' acetylator rabbits re- vealed that, for certain substrates, extrahepatic N-acetyltransferase contributes signifi- cantly to the total acetylating capacity of the individual. The possible significance and applicability ofthese findings to drugmetabolism and acetylation polymorphism in man is discussed. Liver N-acetyltransferase catalyses the acetylation purified by the same procedure and their pH charac- of a number of commonly used drugs and foreign teristics, heat stabilities, kinetic properties, substrate compounds such as isoniazid, sulphamethazine, specificities and reaction mechanisms are indis- sulphadiazine, p-aminobenzoic acid, diamino- tinguishable. -

Chapter 13 Reactions of Arenes Electrophilic Aromatic Substitution
CH. 13 Chapter 13 Reactions of Arenes Electrophilic Aromatic Substitution Electrophiles add to aromatic rings in a fashion somewhat similar to the addition of electrophiles to alkenes. Recall: R3 R4 E Y E Y C C + E Y R1 C C R4 R1 C C R4 − R2 R1 δ+ δ R2 R3 R2 R3 In aromatic rings, however, we see substitution of one of the benzene ring hydrogens for an electrophile. H E + E Y + Y H δ+ δ− The mechanism is the same regardless of the electrophile. It involves two steps: (1) Addition of the electrophile to form a high-energy carbocation. (2) Elimination of the proton to restore the aromatic ring system. H H H H slow E Y + Y step 1 + E δ+ δ− high energy arenium ion H H step 2 fast E E + Y + Y H The first step is the slow step since the aromaticity of the benzene ring system is destroyed on formation of the arenium ion intermediate. This is a high energy species but it is stabilized by resonance with the remaining two double bonds. The second step is very fast since it restores the aromatic stabilization. 1 CH. 13 H H H H H H E E E There are five electrophilic aromatic substitution reactions that we will study. (1) Nitration H NO2 H2SO4 + HNO3 (2) Sulfonation H SO3H + H2SO4 (3) Halogenation with bromine or chlorine H X FeX3 X = Br, Cl + X2 (4) Friedel-Crafts Alkylation H R AlX + RX 3 (5) Friedel-Crafts Acylation O H O C AlX3 R + Cl C R 2 CH. -

The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc Oncogenesis
The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis By Yuting Sun This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy at the University of New South Wales Children’s Cancer Institute Australia for Medical Research School of Women’s and Children’s Health, Faculty of Medicine University of New South Wales Australia August 2014 PLEASE TYPE THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Sun First name: Yuting Other name/s: Abbreviation for degree as given in the University calendar: PhD School : School of·Women's and Children's Health Faculty: Faculty of Medicine Title: The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis. Abstract 350 words maximum: (PLEASE TYPE) N-Myc Induces neuroblastoma by regulating the expression of target genes and proteins, and N-Myc protein is degraded by Fbxw7 and NEDD4 and stabilized by Aurora A. The class lla histone deacetylase HDAC5 suppresses gene transcription, and blocks myoblast and leukaemia cell differentiation. While histone H3 lysine 4 (H3K4) trimethylation at target gene promoters is a pre-requisite for Myc· induced transcriptional activation, WDRS, as a histone H3K4 methyltransferase presenter, is required for H3K4 methylation and transcriptional activation mediated by a histone H3K4 methyltransferase complex. Here, I investigated the roles of HDAC5 and WDR5 in N-Myc overexpressing neuroblastoma. I have found that N-Myc upregulates HDAC5 protein expression, and that HDAC5 represses NEDD4 gene expression, increases Aurora A gene expression and consequently upregulates N-Myc protein expression in neuroblastoma cells. -

The Histone Deacetylase HDA15 Interacts with MAC3A and MAC3B to Regulate Intron
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.386672; this version posted November 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The histone deacetylase HDA15 interacts with MAC3A and MAC3B to regulate intron 2 retention of ABA-responsive genes 3 4 Yi-Tsung Tu1#, Chia-Yang Chen1#, Yi-Sui Huang1, Ming-Ren Yen2, Jo-Wei Allison Hsieh2,3, 5 Pao-Yang Chen2,3*and Keqiang Wu1* 6 7 1Institute of Plant Biology, National Taiwan University, Taipei 10617, Taiwan 8 2 Institute of Plant and Microbial Biology, Academia Sinica, Taipei 11529, Taiwan 9 3 Genome and Systems Biology Degree Program, Academia Sinica and National Taiwan 10 University, Taipei, Taiwan 11 12 # These authors contributed equally to this work. 13 * Corresponding authors: Keqiang Wu ([email protected], +886-2-3366-4546) and Pao-Yang 14 Chen ([email protected], +886-2-2787-1140) 15 16 Short title: HDA15 and MAC3A/MAC3B coregulate intron retention 17 One sentence summary: HDA15 and MAC3A/MAC3B coregulate intron retention and reduce 18 the histone acetylation level of the genomic regions near ABA-responsive retained introns. 19 20 The author responsible for distribution of materials integral to the findings presented in this 21 article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) 22 is: Keqiang Wu ([email protected]) 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.386672; this version posted November 17, 2020. -

KAT5 Acetylates Cgas to Promote Innate Immune Response to DNA Virus
KAT5 acetylates cGAS to promote innate immune response to DNA virus Ze-Min Songa, Heng Lina, Xue-Mei Yia, Wei Guoa, Ming-Ming Hua, and Hong-Bing Shua,1 aDepartment of Infectious Diseases, Zhongnan Hospital of Wuhan University, Frontier Science Center for Immunology and Metabolism, Medical Research Institute, Wuhan University, 430071 Wuhan, China Edited by Adolfo Garcia-Sastre, Icahn School of Medicine at Mount Sinai, New York, NY, and approved July 30, 2020 (received for review December 19, 2019) The DNA sensor cGMP-AMP synthase (cGAS) senses cytosolic mi- suppress its enzymatic activity (15). It has also been shown that crobial or self DNA to initiate a MITA/STING-dependent innate im- the NUD of cGAS is critically involved in its optimal DNA- mune response. cGAS is regulated by various posttranslational binding (16), phase-separation (7), and subcellular locations modifications at its C-terminal catalytic domain. Whether and (17). However, whether and how the NUD of cGAS is regulated how its N-terminal unstructured domain is regulated by posttrans- remains unknown. lational modifications remain unknown. We identified the acetyl- The lysine acetyltransferase 5 (KAT5) is a catalytic subunit of transferase KAT5 as a positive regulator of cGAS-mediated innate the highly conserved NuA4 acetyltransferase complex, which immune signaling. Overexpression of KAT5 potentiated viral- plays critical roles in DNA damage repair, p53-mediated apo- DNA–triggered transcription of downstream antiviral genes, whereas ptosis, HIV-1 transcription, and autophagy (18–21). Although a KAT5 deficiency had the opposite effects. Mice with inactivated KAT5 has been investigated mostly as a transcriptional regula- Kat5 exhibited lower levels of serum cytokines in response to DNA tor, there is increasing evidence that KAT5 also acts as a key virus infection, higher viral titers in the brains, and more susceptibility regulator in signal transduction pathways by targeting nonhis- to DNA-virus–induced death. -
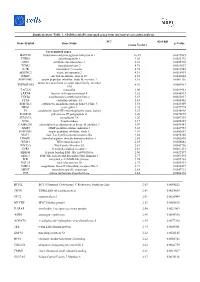
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Distinct Contributions of DNA Methylation and Histone Acetylation to the Genomic Occupancy of Transcription Factors
Downloaded from genome.cshlp.org on October 8, 2021 - Published by Cold Spring Harbor Laboratory Press Research Distinct contributions of DNA methylation and histone acetylation to the genomic occupancy of transcription factors Martin Cusack,1 Hamish W. King,2 Paolo Spingardi,1 Benedikt M. Kessler,3 Robert J. Klose,2 and Skirmantas Kriaucionis1 1Ludwig Institute for Cancer Research, University of Oxford, Oxford, OX3 7DQ, United Kingdom; 2Department of Biochemistry, University of Oxford, Oxford, OX1 3QU, United Kingdom; 3Target Discovery Institute, University of Oxford, Oxford, OX3 7FZ, United Kingdom Epigenetic modifications on chromatin play important roles in regulating gene expression. Although chromatin states are often governed by multilayered structure, how individual pathways contribute to gene expression remains poorly under- stood. For example, DNA methylation is known to regulate transcription factor binding but also to recruit methyl-CpG binding proteins that affect chromatin structure through the activity of histone deacetylase complexes (HDACs). Both of these mechanisms can potentially affect gene expression, but the importance of each, and whether these activities are inte- grated to achieve appropriate gene regulation, remains largely unknown. To address this important question, we measured gene expression, chromatin accessibility, and transcription factor occupancy in wild-type or DNA methylation-deficient mouse embryonic stem cells following HDAC inhibition. We observe widespread increases in chromatin accessibility at ret- rotransposons when HDACs are inhibited, and this is magnified when cells also lack DNA methylation. A subset of these elements has elevated binding of the YY1 and GABPA transcription factors and increased expression. The pronounced ad- ditive effect of HDAC inhibition in DNA methylation–deficient cells demonstrates that DNA methylation and histone deacetylation act largely independently to suppress transcription factor binding and gene expression. -

Fatty Acid Biosynthesis
BI/CH 422/622 ANABOLISM OUTLINE: Photosynthesis Carbon Assimilation – Calvin Cycle Carbohydrate Biosynthesis in Animals Gluconeogenesis Glycogen Synthesis Pentose-Phosphate Pathway Regulation of Carbohydrate Metabolism Anaplerotic reactions Biosynthesis of Fatty Acids and Lipids Fatty Acids contrasts Diversification of fatty acids location & transport Eicosanoids Synthesis Prostaglandins and Thromboxane acetyl-CoA carboxylase Triacylglycerides fatty acid synthase ACP priming Membrane lipids 4 steps Glycerophospholipids Control of fatty acid metabolism Sphingolipids Isoprene lipids: Cholesterol ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1 ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1. Biosynthesis of fatty acids 2. Regulation of fatty acid degradation and synthesis 3. Assembly of fatty acids into triacylglycerol and phospholipids 4. Metabolism of isoprenes a. Ketone bodies and Isoprene biosynthesis b. Isoprene polymerization i. Cholesterol ii. Steroids & other molecules iii. Regulation iv. Role of cholesterol in human disease ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Degradation Synthesis Ketone body Isoprene Utilization Biosynthesis 2 Catabolism Fatty Acid Biosynthesis Anabolism • Contrast with Sugars – Lipids have have hydro-carbons not carbo-hydrates – more reduced=more energy – Long-term storage vs short-term storage – Lipids are essential for structure in ALL organisms: membrane phospholipids • Catabolism of fatty acids –produces acetyl-CoA –produces reducing -

Phylogenetic Analysis, Subcellular Localization, and Expression
BMC Plant Biology BioMed Central Research article Open Access Phylogenetic analysis, subcellular localization, and expression patterns of RPD3/HDA1 family histone deacetylases in plants Malona V Alinsug, Chun-Wei Yu and Keqiang Wu* Address: Institute of Plant Biology, College of Life Science, National Taiwan University, Taipei, Taiwan Email: Malona V Alinsug - [email protected]; Chun-Wei Yu - [email protected]; Keqiang Wu* - [email protected] * Corresponding author Published: 28 March 2009 Received: 26 November 2008 Accepted: 28 March 2009 BMC Plant Biology 2009, 9:37 doi:10.1186/1471-2229-9-37 This article is available from: http://www.biomedcentral.com/1471-2229/9/37 © 2009 Alinsug et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Although histone deacetylases from model organisms have been previously identified, there is no clear basis for the classification of histone deacetylases under the RPD3/ HDA1 superfamily, particularly on plants. Thus, this study aims to reconstruct a phylogenetic tree to determine evolutionary relationships between RPD3/HDA1 histone deacetylases from six different plants representing dicots with Arabidopsis thaliana, Populus trichocarpa, and Pinus taeda, monocots with Oryza sativa and Zea mays, and the lower plants with Physcomitrella patens. Results: Sixty two histone deacetylases of RPD3/HDA1 family from the six plant species were phylogenetically analyzed to determine corresponding orthologues. Three clusters were formed separating Class I, Class II, and Class IV.