Biosynthesis of Natural Products Containing Β-Amino Acids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Report of the Advisory Group to Recommend Priorities for the IARC Monographs During 2020–2024
IARC Monographs on the Identification of Carcinogenic Hazards to Humans Report of the Advisory Group to Recommend Priorities for the IARC Monographs during 2020–2024 Report of the Advisory Group to Recommend Priorities for the IARC Monographs during 2020–2024 CONTENTS Introduction ................................................................................................................................... 1 Acetaldehyde (CAS No. 75-07-0) ................................................................................................. 3 Acrolein (CAS No. 107-02-8) ....................................................................................................... 4 Acrylamide (CAS No. 79-06-1) .................................................................................................... 5 Acrylonitrile (CAS No. 107-13-1) ................................................................................................ 6 Aflatoxins (CAS No. 1402-68-2) .................................................................................................. 8 Air pollutants and underlying mechanisms for breast cancer ....................................................... 9 Airborne gram-negative bacterial endotoxins ............................................................................. 10 Alachlor (chloroacetanilide herbicide) (CAS No. 15972-60-8) .................................................. 10 Aluminium (CAS No. 7429-90-5) .............................................................................................. 11 -

QM/MM) Study on Ornithine Cyclodeaminase (OCD): a Tale of Two Iminiums
Int. J. Mol. Sci. 2012, 13, 12994-13011; doi:10.3390/ijms131012994 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article A Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) Study on Ornithine Cyclodeaminase (OCD): A Tale of Two Iminiums Bogdan F. Ion, Eric A. C. Bushnell, Phil De Luna and James W. Gauld * Department of Chemistry and Biochemistry, University of Windsor, Windsor, ON N9B 3P4, Canada; E-Mails: [email protected] (B.F.I.); [email protected] (E.A.C.B.); [email protected] (P.D.L.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-519-253-3000; Fax: +1-519-973-7098. Received: 10 September 2012; in revised form: 27 September 2012 / Accepted: 27 September 2012 / Published: 11 October 2012 Abstract: Ornithine cyclodeaminase (OCD) is an NAD+-dependent deaminase that is found in bacterial species such as Pseudomonas putida. Importantly, it catalyzes the direct conversion of the amino acid L-ornithine to L-proline. Using molecular dynamics (MD) and a hybrid quantum mechanics/molecular mechanics (QM/MM) method in the ONIOM formalism, the catalytic mechanism of OCD has been examined. The rate limiting step is calculated to be the initial step in the overall mechanism: hydride transfer from the + + L-ornithine’s Cα–H group to the NAD cofactor with concomitant formation of a Cα=NH2 Schiff base with a barrier of 90.6 kJ mol−1. Importantly, no water is observed within the + active site during the MD simulations suitably positioned to hydrolyze the Cα=NH2 intermediate to form the corresponding carbonyl. -

Cyanobacterial Peptide Toxins
CYANOBACTERIAL PEPTIDE TOXINS CYANOBACTERIAL PEPTIDE TOXINS 1. Exposure data 1.1 Introduction Cyanobacteria, also known as blue-green algae, are widely distributed in fresh, brackish and marine environments, in soil and on moist surfaces. They are an ancient group of prokaryotic organisms that are found all over the world in environments as diverse as Antarctic soils and volcanic hot springs, often where no other vegetation can exist (Knoll, 2008). Cyanobacteria are considered to be the organisms responsible for the early accumulation of oxygen in the earth’s atmosphere (Knoll, 2008). The name ‘blue- green’ algae derives from the fact that these organisms contain a specific pigment, phycocyanin, which gives many species a slightly blue-green appearance. Cyanobacterial metabolites can be lethally toxic to wildlife, domestic livestock and even humans. Cyanotoxins fall into three broad groups of chemical structure: cyclic peptides, alkaloids and lipopolysaccharides. Table 1.1 gives an overview of the specific toxic substances within these broad groups that are produced by different genera of cyanobacteria together, with their primary target organs in mammals. However, not all cyanobacterial blooms are toxic and neither are all strains within one species. Toxic and non-toxic strains show no predictable difference in appearance and, therefore, physicochemical, biochemical and biological methods are essential for the detection of cyanobacterial toxins. The most frequently reported cyanobacterial toxins are cyclic heptapeptide toxins known as microcystins which can be isolated from several species of the freshwater genera Microcystis , Planktothrix ( Oscillatoria ), Anabaena and Nostoc . More than 70 structural variants of microcystins are known. A structurally very similar class of cyanobacterial toxins is nodularins ( < 10 structural variants), which are cyclic pentapeptide hepatotoxins that are found in the brackish-water cyanobacterium Nodularia . -

Identification of the Toxic Pentapeptide Nodularin in A
A tica nal eu yt c ic a a m A r a c t h a P Pacheco et al., Pharm Anal Acta 2016, 7:5 Pharmaceutica Analytica Acta DOI: 10.4172/2153-2435.1000479 ISSN: 2153-2435 Research Article Open Access Identification of the Toxic Pentapeptide Nodularin in a Cyanobacterial Bloom in a Shrimp Farm in South American Atlantic Coast Pacheco LA1,3, Kunrath N1, Costa CM1,4, Costa LDF1, Foes GK2, Wasielesky W2 and Yunes JS1* 1Laboratory of Cyanobacteria and Phycotoxins, Institute of Oceanography, Federal University of Rio Grande, RS, Brazil 2Aquaculture Marine Station (EMA), Institute of Oceanography, Federal University of Rio Grande, RS, Brazil 3Post Graduate Program in Physical, Chemical and Geological Oceanography , Institute of Oceanography, Federal University of Rio Grande, RS, Brazil 4Post Graduate Program in Aquaculture, Institute of Oceanography, Federal University of Rio Grande, RS, Brazil *Corresponding author: Yunes JS, Laboratório de Cianobactérias e Ficotoxinas, IOFURG, Universidade Federal do Rio Grande, 96.203-270 - Rio Grande, RS, Brazil, Tel: +55 53 32336737; E-mail: [email protected] Received date: Apr 28, 2016; Accepted date: May 23, 2016; Published date: May 25, 2016 Copyright: © 2016 Pacheco LA et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Since 2010, blooms of the brackish cyanobacteria Nodularia spumigena are recurrent in the shrimp growth tanks of the Marine Aquaculture Station during summer in Southern Brazil. Cyanobacterial growth led to a decrease in the white shrimp Litopenaeus vannamei productivity. -

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

Cyanobacteria 101 (And Why You Need to Know More)
Cyanobacteria 101 (and why you need to know more) Barry H. Rosen, Ph. D. Office of the Southeast Regional Director (CFLWSC) Orlando, FL [email protected] 407-803-5508 algae algae 32 million cyanobacteria cyanobacteria 2.9 million HABs 372 K HABs microcystin 314 K saxitoxin 195 K microcystin paralytic 143 K amnesic 56 K saxitoxin cyanotoxins 48 K *paralytic shellfish poisoning (PSP) amnesic shellfish poisoning (ASP) cyanotoxins *coastal & marine Cyanobacteria (aka blue- green algae; cyanoHABs) •gram negative bacteria •pigments in thylakoids WhereWhere are cyanobacteriaare they a problem? a problem? Lakes, reservoirs, rivers, streams, wetlands Estuaries and coastal systems Marine systems CyanoHABs NOAA, OSU, SeaGrant Why are we concerned about cyanoHABs? Toxicity Hypoxia Taste and odors Aesthetics So why do we care about them? Some produce cyanobacteria toxins Cyanotoxins Hepatotoxins Disrupt proteins that keep microcystin (90+ variants) the liver functioning, may nodularin act slowly (days to weeks) cylindrospermopsin Neurotoxins anatoxin -a Cause rapid paralysis of anatoxin -a (s) skeletal and respiratory saxitoxin muscles (minutes) neosaxitoxin Dermatotoxins lyngbyatoxin Produce rashes and other skin reactions, usually within a day (hours) b-N-methylamino-L-alanine BMAA Neurological: linked to ALS Cyanotoxins are highly potent Compounds & LD50 (ug/kg) Saxitoxin 9 Ricin 0.02 Anatoxin-a(s) 20 Cobra toxin 20 Microcystin LR 50 Curare 500 Anatoxin-a 200-250 Strychnine 2000 Nodularin 50 Cylindrospermopsins 200 How common are toxic blooms? -
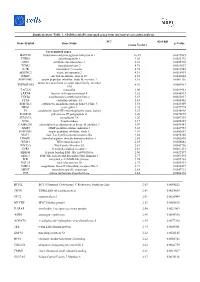
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

The Neurotoxin Β-N-Methylamino-L-Alanine (BMAA)
The neurotoxin β-N-methylamino-L-alanine (BMAA) Sources, bioaccumulation and extraction procedures Sandra Ferreira Lage ©Sandra Ferreira Lage, Stockholm University 2016 Cover image: Cyanobacteria, diatoms and dinoflagellates microscopic pictures taken by Sandra Ferreira Lage ISBN 978-91-7649-455-4 Printed in Sweden by Holmbergs, Malmö 2016 Distributor: Department of Ecology, Environment and Plant Sciences, Stockholm University “Sinto mais longe o passado, sinto a saudade mais perto.” Fernando Pessoa, 1914. Abstract β-methylamino-L-alanine (BMAA) is a neurotoxin linked to neurodegeneration, which is manifested in the devastating human diseases amyotrophic lateral sclerosis, Alzheimer’s and Parkinson’s disease. This neurotoxin is known to be produced by almost all tested species within the cyanobacterial phylum including free living as well as the symbiotic strains. The global distribution of the BMAA producers ranges from a terrestrial ecosystem on the Island of Guam in the Pacific Ocean to an aquatic ecosystem in Northern Europe, the Baltic Sea, where annually massive surface blooms occur. BMAA had been shown to accumulate in the Baltic Sea food web, with highest levels in the bottom dwelling fish-species as well as in mollusks. One of the aims of this thesis was to test the bottom-dwelling bioaccumulation hy- pothesis by using a larger number of samples allowing a statistical evaluation. Hence, a large set of fish individuals from the lake Finjasjön, were caught and the BMAA concentrations in different tissues were related to the season of catching, fish gender, total weight and species. The results reveal that fish total weight and fish species were positively correlated with BMAA concentration in the fish brain. -

Physicochemical Properties of Organic Medicinal Agents
Principles of Drug Action 1, Spring 2005, Esters ESTERS AND RELATED CARBOXYLIC ACID DERIVATIVES Jack DeRuiter I. Structure and Preparation Esters are derivatives of carboxylic acids that arise via replacement of the hydroxyl (OH) portion of the acid COOH function with an "ether" moiety (-OR): O O H C C O C O Acid Ester Note that replacement of the acid OH group with an "ether" moiety removes the acidic function from the parent structure (acid) resulting in the formation of non-acidic (neutral, but somewhat polar) compounds (esters). Esters can be sub-classified based on their general structure as aliphatic, aromatic or cyclic (called "lactones") as illustrated by the examples below: O O CH2CH3 CH2CH3 O CH3 O O O Aliphatic Ester Aromatic Ester Cyclic Ester (Lactone) A variety of methods have been developed for the preparation of esters. Most of these methods involve reaction of an alcohol with an "activated carboxylic acid" compound (i.e. acid chloride): O O H C C X OC C O X- Ester "Activated" acid (X=Cl) Alcohol (Electrophile) (Nucleophile) The ester functionality does not introduce a center of asymmetry and thus optical and geometric isomerism does not result from the presence of this functional group. The ester functionality (the carbonyl and ether oxygen) is composed of an sp2 hybridized carbon so it cannot be chiral, and since there is free rotation about the ether bond geometric isomerism also is not possible at the sp2 center. 1 Principles of Drug Action 1, Spring 2005, Esters II. Solubility of Esters Esters contain carbonyl (C=O) and ether (O-C) dipoles arising from covalent bonding between electronegative oxygen atoms and electronically neutral carbon atoms. -

ORGANIC CHEMICAL TOXICOLOGY of FISHES This Is Volume 33 in The
ORGANIC CHEMICAL TOXICOLOGY OF FISHES This is Volume 33 in the FISH PHYSIOLOGY series Edited by Anthony P. Farrell and Colin J. Brauner Honorary Editors: William S. Hoar and David J. Randall A complete list of books in this series appears at the end of the volume ORGANIC CHEMICAL TOXICOLOGY OF FISHES Edited by KEITH B. TIERNEY Department of Biological Sciences University of Alberta Edmonton, Alberta Canada ANTHONY P. FARRELL Department of Zoology, and Faculty of Land and Food Systems The University of British Columbia Vancouver, British Columbia Canada COLIN J. BRAUNER Department of Zoology The University of British Columbia Vancouver, British Columbia Canada AMSTERDAM BOSTON HEIDELBERG LONDON NEW YORK OXFORD PARIS SAN DIEGO SAN FRANCISCO SINGAPORE SYDNEY TOKYO Academic Press is an imprint of Elsevier Academic Press is an imprint of Elsevier 32 Jamestown Road, London NW1 7BY, UK 225 Wyman Street, Waltham, MA 02451, USA 525 B Street, Suite 1800, San Diego, CA 92101-4495, USA Copyright r 2014 Elsevier Inc. All rights reserved The cover illustrates the diversity of effects an example synthetic organic water pollutant can have on fish. The chemical shown is 2,4-D, an herbicide that can be found in streams near urbanization and agriculture. The fish shown is one that can live in such streams: rainbow trout (Oncorhynchus mykiss). The effect shown on the left is the ability of 2,4-D (yellow line) to stimulate olfactory sensory neurons vs. control (black line) (measured as an electro- olfactogram; EOG). The effect shown on the right is the ability of 2,4-D to induce the expression of an egg yolk precursor protein (vitellogenin) in male fish. -

Biochemical Characterization and Comparison of Aspartylglucosaminidases Secreted in Venom of the Parasitoid Wasps Asobara Tabida and Leptopilina Heterotoma
RESEARCH ARTICLE Biochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma Quentin Coulette1, SeÂverine Lemauf2, Dominique Colinet2, Geneviève PreÂvost1, Caroline Anselme1, Marylène Poirie 2, Jean-Luc Gatti2* a1111111111 1 Unite ªEcologie et Dynamique des Systèmes AnthropiseÂsº (EDYSAN, FRE 3498 CNRS-UPJV), Universite de Picardie Jules Verne, Amiens, France, 2 Universite CoÃte d'Azur, INRA, CNRS, ISA, Sophia Antipolis, a1111111111 France a1111111111 a1111111111 * [email protected] a1111111111 Abstract Aspartylglucosaminidase (AGA) is a low-abundance intracellular enzyme that plays a key OPEN ACCESS role in the last stage of glycoproteins degradation, and whose deficiency leads to human Citation: Coulette Q, Lemauf S, Colinet D, PreÂvost aspartylglucosaminuria, a lysosomal storage disease. Surprisingly, high amounts of AGA- G, Anselme C, Poirie M, et al. (2017) Biochemical characterization and comparison of like proteins are secreted in the venom of two phylogenetically distant hymenopteran para- aspartylglucosaminidases secreted in venom of the sitoid wasp species, Asobara tabida (Braconidae) and Leptopilina heterotoma (Cynipidae). parasitoid wasps Asobara tabida and Leptopilina These venom AGAs have a similar domain organization as mammalian AGAs. They share heterotoma. PLoS ONE 12(7): e0181940. https:// with them key residues for autocatalysis and activity, and the mature - and -subunits also doi.org/10.1371/journal.pone.0181940 α β form an (αβ)2 structure in solution. Interestingly, only one of these AGAs subunits (α for Editor: Erjun Ling, Institute of Plant Physiology and AtAGA and for LhAGA) is glycosylated instead of the two subunits for lysosomal human Ecology Shanghai Institutes for Biological β Sciences, CHINA AGA (hAGA), and these glycosylations are partially resistant to PGNase F treatment. -

Functional Characterization of an Ornithine Cyclodeaminase-Like Protein of Arabidopsis Thaliana Sandeep Sharma, Suhas Shinde and Paul E Verslues*
Sharma et al. BMC Plant Biology 2013, 13:182 http://www.biomedcentral.com/1471-2229/13/182 RESEARCH ARTICLE Open Access Functional characterization of an ornithine cyclodeaminase-like protein of Arabidopsis thaliana Sandeep Sharma, Suhas Shinde and Paul E Verslues* Abstract Background: In plants, proline synthesis occurs by two enzymatic steps starting from glutamate as a precursor. Some bacteria, including bacteria such as Agrobacterium rhizogenes have an Ornithine Cyclodeaminase (OCD) which can synthesize proline in a single step by deamination of ornithine. In A. rhizogenes, OCD is one of the genes transferred to the plant genome during the transformation process and plants expressing A. rhizogenes OCD have developmental phenotypes. One nuclear encoded gene of Arabidopsis thaliana has recently been annotated as an OCD (OCD-like; referred to here as AtOCD) but nothing is known of its function. As proline metabolism contributes to tolerance of low water potential during drought, it is of interest to determine if AtOCD affects proline accumulation or low water potential tolerance. Results: Expression of AtOCD was induced by low water potential stress and by exogenous proline, but not by the putative substrate ornithine. The AtOCD protein was plastid localized. T-DNA mutants of atocd and AtOCD RNAi plants had approximately 15% higher proline accumulation at low water potential while p5cs1-4/atocd double mutants had 40% higher proline than p5cs1 at low water potential but no change in proline metabolism gene expression which could directly explain the higher proline level. AtOCD overexpression did not affect proline accumulation. Enzymatic assays with bacterially expressed AtOCD or AtOCD purified from AtOCD:Flag transgenic plants did not detect any activity using ornithine, proline or several other amino acids as substrates.