Identification of Novel Small-Molecule Histone Deacetylase Inhibitors By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

Interplay Between Epigenetics and Metabolism in Oncogenesis: Mechanisms and Therapeutic Approaches
OPEN Oncogene (2017) 36, 3359–3374 www.nature.com/onc REVIEW Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches CC Wong1, Y Qian2,3 and J Yu1 Epigenetic and metabolic alterations in cancer cells are highly intertwined. Oncogene-driven metabolic rewiring modifies the epigenetic landscape via modulating the activities of DNA and histone modification enzymes at the metabolite level. Conversely, epigenetic mechanisms regulate the expression of metabolic genes, thereby altering the metabolome. Epigenetic-metabolomic interplay has a critical role in tumourigenesis by coordinately sustaining cell proliferation, metastasis and pluripotency. Understanding the link between epigenetics and metabolism could unravel novel molecular targets, whose intervention may lead to improvements in cancer treatment. In this review, we summarized the recent discoveries linking epigenetics and metabolism and their underlying roles in tumorigenesis; and highlighted the promising molecular targets, with an update on the development of small molecule or biologic inhibitors against these abnormalities in cancer. Oncogene (2017) 36, 3359–3374; doi:10.1038/onc.2016.485; published online 16 January 2017 INTRODUCTION metabolic genes have also been identified as driver genes It has been appreciated since the early days of cancer research mutated in some cancers, such as isocitrate dehydrogenase 1 16 17 that the metabolic profiles of tumor cells differ significantly from and 2 (IDH1/2) in gliomas and acute myeloid leukemia (AML), 18 normal cells. Cancer cells have high metabolic demands and they succinate dehydrogenase (SDH) in paragangliomas and fuma- utilize nutrients with an altered metabolic program to support rate hydratase (FH) in hereditary leiomyomatosis and renal cell 19 their high proliferative rates and adapt to the hostile tumor cancer (HLRCC). -

The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc Oncogenesis
The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis By Yuting Sun This thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy at the University of New South Wales Children’s Cancer Institute Australia for Medical Research School of Women’s and Children’s Health, Faculty of Medicine University of New South Wales Australia August 2014 PLEASE TYPE THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Sun First name: Yuting Other name/s: Abbreviation for degree as given in the University calendar: PhD School : School of·Women's and Children's Health Faculty: Faculty of Medicine Title: The Roles of Histone Deacetylase 5 and the Histone Methyltransferase Adaptor WDR5 in Myc oncogenesis. Abstract 350 words maximum: (PLEASE TYPE) N-Myc Induces neuroblastoma by regulating the expression of target genes and proteins, and N-Myc protein is degraded by Fbxw7 and NEDD4 and stabilized by Aurora A. The class lla histone deacetylase HDAC5 suppresses gene transcription, and blocks myoblast and leukaemia cell differentiation. While histone H3 lysine 4 (H3K4) trimethylation at target gene promoters is a pre-requisite for Myc· induced transcriptional activation, WDRS, as a histone H3K4 methyltransferase presenter, is required for H3K4 methylation and transcriptional activation mediated by a histone H3K4 methyltransferase complex. Here, I investigated the roles of HDAC5 and WDR5 in N-Myc overexpressing neuroblastoma. I have found that N-Myc upregulates HDAC5 protein expression, and that HDAC5 represses NEDD4 gene expression, increases Aurora A gene expression and consequently upregulates N-Myc protein expression in neuroblastoma cells. -

The Histone Deacetylase HDA15 Interacts with MAC3A and MAC3B to Regulate Intron
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.386672; this version posted November 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The histone deacetylase HDA15 interacts with MAC3A and MAC3B to regulate intron 2 retention of ABA-responsive genes 3 4 Yi-Tsung Tu1#, Chia-Yang Chen1#, Yi-Sui Huang1, Ming-Ren Yen2, Jo-Wei Allison Hsieh2,3, 5 Pao-Yang Chen2,3*and Keqiang Wu1* 6 7 1Institute of Plant Biology, National Taiwan University, Taipei 10617, Taiwan 8 2 Institute of Plant and Microbial Biology, Academia Sinica, Taipei 11529, Taiwan 9 3 Genome and Systems Biology Degree Program, Academia Sinica and National Taiwan 10 University, Taipei, Taiwan 11 12 # These authors contributed equally to this work. 13 * Corresponding authors: Keqiang Wu ([email protected], +886-2-3366-4546) and Pao-Yang 14 Chen ([email protected], +886-2-2787-1140) 15 16 Short title: HDA15 and MAC3A/MAC3B coregulate intron retention 17 One sentence summary: HDA15 and MAC3A/MAC3B coregulate intron retention and reduce 18 the histone acetylation level of the genomic regions near ABA-responsive retained introns. 19 20 The author responsible for distribution of materials integral to the findings presented in this 21 article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) 22 is: Keqiang Wu ([email protected]) 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.17.386672; this version posted November 17, 2020. -
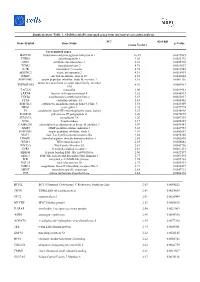
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Distinct Contributions of DNA Methylation and Histone Acetylation to the Genomic Occupancy of Transcription Factors
Downloaded from genome.cshlp.org on October 8, 2021 - Published by Cold Spring Harbor Laboratory Press Research Distinct contributions of DNA methylation and histone acetylation to the genomic occupancy of transcription factors Martin Cusack,1 Hamish W. King,2 Paolo Spingardi,1 Benedikt M. Kessler,3 Robert J. Klose,2 and Skirmantas Kriaucionis1 1Ludwig Institute for Cancer Research, University of Oxford, Oxford, OX3 7DQ, United Kingdom; 2Department of Biochemistry, University of Oxford, Oxford, OX1 3QU, United Kingdom; 3Target Discovery Institute, University of Oxford, Oxford, OX3 7FZ, United Kingdom Epigenetic modifications on chromatin play important roles in regulating gene expression. Although chromatin states are often governed by multilayered structure, how individual pathways contribute to gene expression remains poorly under- stood. For example, DNA methylation is known to regulate transcription factor binding but also to recruit methyl-CpG binding proteins that affect chromatin structure through the activity of histone deacetylase complexes (HDACs). Both of these mechanisms can potentially affect gene expression, but the importance of each, and whether these activities are inte- grated to achieve appropriate gene regulation, remains largely unknown. To address this important question, we measured gene expression, chromatin accessibility, and transcription factor occupancy in wild-type or DNA methylation-deficient mouse embryonic stem cells following HDAC inhibition. We observe widespread increases in chromatin accessibility at ret- rotransposons when HDACs are inhibited, and this is magnified when cells also lack DNA methylation. A subset of these elements has elevated binding of the YY1 and GABPA transcription factors and increased expression. The pronounced ad- ditive effect of HDAC inhibition in DNA methylation–deficient cells demonstrates that DNA methylation and histone deacetylation act largely independently to suppress transcription factor binding and gene expression. -

Biochemical Characterization and Comparison of Aspartylglucosaminidases Secreted in Venom of the Parasitoid Wasps Asobara Tabida and Leptopilina Heterotoma
RESEARCH ARTICLE Biochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma Quentin Coulette1, SeÂverine Lemauf2, Dominique Colinet2, Geneviève PreÂvost1, Caroline Anselme1, Marylène Poirie 2, Jean-Luc Gatti2* a1111111111 1 Unite ªEcologie et Dynamique des Systèmes AnthropiseÂsº (EDYSAN, FRE 3498 CNRS-UPJV), Universite de Picardie Jules Verne, Amiens, France, 2 Universite CoÃte d'Azur, INRA, CNRS, ISA, Sophia Antipolis, a1111111111 France a1111111111 a1111111111 * [email protected] a1111111111 Abstract Aspartylglucosaminidase (AGA) is a low-abundance intracellular enzyme that plays a key OPEN ACCESS role in the last stage of glycoproteins degradation, and whose deficiency leads to human Citation: Coulette Q, Lemauf S, Colinet D, PreÂvost aspartylglucosaminuria, a lysosomal storage disease. Surprisingly, high amounts of AGA- G, Anselme C, Poirie M, et al. (2017) Biochemical characterization and comparison of like proteins are secreted in the venom of two phylogenetically distant hymenopteran para- aspartylglucosaminidases secreted in venom of the sitoid wasp species, Asobara tabida (Braconidae) and Leptopilina heterotoma (Cynipidae). parasitoid wasps Asobara tabida and Leptopilina These venom AGAs have a similar domain organization as mammalian AGAs. They share heterotoma. PLoS ONE 12(7): e0181940. https:// with them key residues for autocatalysis and activity, and the mature - and -subunits also doi.org/10.1371/journal.pone.0181940 α β form an (αβ)2 structure in solution. Interestingly, only one of these AGAs subunits (α for Editor: Erjun Ling, Institute of Plant Physiology and AtAGA and for LhAGA) is glycosylated instead of the two subunits for lysosomal human Ecology Shanghai Institutes for Biological β Sciences, CHINA AGA (hAGA), and these glycosylations are partially resistant to PGNase F treatment. -

Pan-Histone Deacetylase Inhibitors Regulate Signaling Pathways
Majumdar et al. BMC Genomics 2012, 13:709 http://www.biomedcentral.com/1471-2164/13/709 RESEARCH ARTICLE Open Access Pan-histone deacetylase inhibitors regulate signaling pathways involved in proliferative and pro-inflammatory mechanisms in H9c2 cells Gipsy Majumdar1, Piyatilake Adris1, Neha Bhargava1, Hao Chen2 and Rajendra Raghow1,2* Abstract Background: We have shown previously that pan-HDAC inhibitors (HDACIs) m-carboxycinnamic acid bis-hydroxamide (CBHA) and trichostatin A (TSA) attenuated cardiac hypertrophy in BALB/c mice by inducing hyper-acetylation of cardiac chromatin that was accompanied by suppression of pro-inflammatory gene networks. However, it was not feasible to determine the precise contribution of the myocytes- and non-myocytes to HDACI-induced gene expression in the intact heart. Therefore, the current study was undertaken with a primary goal of elucidating temporal changes in the transcriptomes of cardiac myocytes exposed to CBHA and TSA. Results: We incubated H9c2 cardiac myocytes in growth medium containing either of the two HDACIs for 6h and 24h and analyzed changes in gene expression using Illumina microarrays. H9c2 cells exposed to TSA for 6h and 24h led to differential expression of 468 and 231 genes, respectively. In contrast, cardiac myocytes incubated with CBHA for 6h and 24h elicited differential expression of 768 and 999 genes, respectively. We analyzed CBHA- and TSA-induced differentially expressed genes by Ingenuity Pathway (IPA), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Core_TF programs and discovered that CBHA and TSA impinged on several common gene networks. Thus, both HDACIs induced a repertoire of signaling kinases (PTEN-PI3K-AKT and MAPK) and transcription factors (Myc, p53, NFkB and HNF4A) representing canonical TGFβ, TNF-α, IFNγ and IL-6 specific networks. -

Histone Deacetylase Inhibitors Synergizes with Catalytic Inhibitors of EZH2 to Exhibit Anti-Tumor Activity in Small Cell Carcinoma of the Ovary, Hypercalcemic Type
Author Manuscript Published OnlineFirst on September 19, 2018; DOI: 10.1158/1535-7163.MCT-18-0348 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Histone deacetylase inhibitors synergizes with catalytic inhibitors of EZH2 to exhibit anti- tumor activity in small cell carcinoma of the ovary, hypercalcemic type Yemin Wang1,2,*, Shary Yuting Chen1,2, Shane Colborne3, Galen Lambert2, Chae Young Shin2, Nancy Dos Santos4, Krystal A. Orlando5, Jessica D. Lang6, William P.D. Hendricks6, Marcel B. Bally4, Anthony N. Karnezis1,2, Ralf Hass7, T. Michael Underhill8, Gregg B. Morin3,9, Jeffrey M. Trent6, Bernard E. Weissman5, David G. Huntsman1,2,10,* 1Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada 2Department of Molecular Oncology, British Columbia Cancer Research Centre, Vancouver, BC, Canada. 3Michael Smith Genome Science Centre, British Columbia Cancer Agency, Vancouver, BC, Canada. 4Department of Experimental Therapeutics, British Columbia Cancer Research Centre, Vancouver, BC, Canada. 5Department of Pathology and Laboratory Medicine and Lineberger Comprehensive Cancer Center, University of North Carolina, Chapel Hill, NC, USA. 6Division of Integrated Cancer Genomics, Translational Genomics Research Institute (TGen), Phoenix, AZ, USA. 7Department of Obstetrics and Gynecology, Hannover Medical School, D-30625 Hannover, Germany. 8Department of Cellular and Physiological Sciences and Biomedical Research Centre, University 1 Downloaded from mct.aacrjournals.org on September 26, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on September 19, 2018; DOI: 10.1158/1535-7163.MCT-18-0348 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Biosynthesis of Natural Products Containing Β-Amino Acids
Natural Product Reports Biosynthesis of natural products containing β -amino acids Journal: Natural Product Reports Manuscript ID: NP-REV-01-2014-000007.R1 Article Type: Review Article Date Submitted by the Author: 21-Apr-2014 Complete List of Authors: Kudo, Fumitaka; Tokyo Institute Of Technology, Department of Chemistry Miyanaga, Akimasa; Tokyo Institute Of Technology, Department of Chemistry Eguchi, T; Tokyo Institute Of Technology, Department of Chemistry and Materials Science Page 1 of 20 Natural Product Reports NPR RSC Publishing REVIEW Biosynthesis of natural products containing βββ- amino acids Cite this: DOI: 10.1039/x0xx00000x Fumitaka Kudo, a Akimasa Miyanaga, a and Tadashi Eguchi *b Received 00th January 2014, We focus here on β-amino acids as components of complex natural products because the presence of β-amino acids Accepted 00th January 2014 produces structural diversity in natural products and provides characteristic architectures beyond that of ordinary DOI: 10.1039/x0xx00000x α-L-amino acids, thus generating significant and unique biological functions in nature. In this review, we first survey the known bioactive β-amino acid-containing natural products including nonribosomal peptides, www.rsc.org/ macrolactam polyketides, and nucleoside-β-amino acid hybrids. Next, the biosynthetic enzymes that form β-amino acids from α-amino acids and de novo synthesis of β-amino acids are summarized. Then, the mechanisms of β- amino acid incorporation into natural products are reviewed. Because it is anticipated that the rational swapping of the β-amino acid moieties with various side chains and stereochemistries by biosynthetic engineering should lead to the creation of novel architectures and bioactive compounds, the accumulation of knowledge regarding β- amino acid-containing natural product biosynthetic machinery could have a significant impact in this field. -

Phosphate Availability and Ectomycorrhizal Symbiosis with Pinus Sylvestris Have Independent Effects on the Paxillus Involutus Transcriptome
This is a repository copy of Phosphate availability and ectomycorrhizal symbiosis with Pinus sylvestris have independent effects on the Paxillus involutus transcriptome. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/168854/ Version: Published Version Article: Paparokidou, C., Leake, J.R. orcid.org/0000-0001-8364-7616, Beerling, D.J. et al. (1 more author) (2020) Phosphate availability and ectomycorrhizal symbiosis with Pinus sylvestris have independent effects on the Paxillus involutus transcriptome. Mycorrhiza. ISSN 0940- 6360 https://doi.org/10.1007/s00572-020-01001-6 Reuse This article is distributed under the terms of the Creative Commons Attribution (CC BY) licence. This licence allows you to distribute, remix, tweak, and build upon the work, even commercially, as long as you credit the authors for the original work. More information and the full terms of the licence here: https://creativecommons.org/licenses/ Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ Mycorrhiza https://doi.org/10.1007/s00572-020-01001-6 ORIGINAL ARTICLE Phosphate availability and ectomycorrhizal symbiosis with Pinus sylvestris have independent effects on the Paxillus involutus transcriptome Christina Paparokidou1 & Jonathan R. Leake1 & David J. Beerling1 & Stephen A. Rolfe1 Received: 16 June 2020 /Accepted: 29 October 2020 # The Author(s) 2020 Abstract Many plant species form symbioses with ectomycorrhizal fungi, which help them forage for limiting nutrients in the soil such as inorganic phosphate (Pi). -

SET-Induced Calcium Signaling and MAPK/ERK Pathway Activation Mediate Dendritic Cell-Like Differentiation of U937 Cells
Leukemia (2005) 19, 1439–1445 & 2005 Nature Publishing Group All rights reserved 0887-6924/05 $30.00 www.nature.com/leu SET-induced calcium signaling and MAPK/ERK pathway activation mediate dendritic cell-like differentiation of U937 cells A Kandilci1 and GC Grosveld1 1Department of Genetics and Tumor Cell Biology, Mail Stop 331, St Jude Children’s Research Hospital, 332 N. Lauderdale, Memphis, TN 38105, USA Human SET, a target of chromosomal translocation in human G1/S transition by allowing cyclin E–CDK2 activity in the leukemia encodes a highly conserved, ubiquitously expressed, presence of p21.11 Second, SET interacts with cyclin B–CDK1.19 nuclear phosphoprotein. SET mediates many functions includ- ing chromatin remodeling, transcription, apoptosis and cell Overexpression of SET inhibits cyclin B-CDK1 activity, which in cycle control. We report that overexpression of SET directs turn, blocks the G2/M transition; this finding suggests a negative 13 differentiation of the human promonocytic cell line U937 along regulatory role for SET in G2/M transition. Overexpression of the dendritic cell (DC) pathway, as cells display typical cell division autoantigen-1 (CDA1), another member of the morphologic changes associated with DC fate and express NAP/SET family, inhibits proliferation and decreases bromo- the DC surface markers CD11b and CD86. Differentiation occurs deoxyuridine uptake in HeLa cells.20 Acidic and basic domains via a calcium-dependent mechanism involving the CaMKII and 20 MAPK/ERK pathways. Similar responses are elicited by inter- of CDA1 show 40% identity and 68% similarity to SET. feron-c (IFN-c) treatment with the distinction that IFN-c signaling We have recently shown that overexpression of SET in the activates the DNA-binding activity of STAT1 whereas SET human promonocytic cell line U937 causes G0/G1 arrest and overexpression does not.