A Gut Pathobiont Synergizes with the Microbiota to Instigate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Co-Infection Associated with Diarrhea in a Colony of <I>Scid
Laboratory Animal Science Vol 48, No 5 Copyright 1998 October 1998 by the American Association for Laboratory Animal Science Helicobacter bilis/Helicobacter rodentium Co-Infection Associated with Diarrhea in a Colony of scid Mice Nirah H. Shomer,* Charles A. Dangler, Robert P. Marini, and James G. Fox† Abstract _ An outbreak of diarrhea spanning 3 months occurred in a breeding colony of scid/Trp53 knockout mice. Approximately a third of the 150 mice were clinically affected, with signs ranging from mucoid or watery diarrhea to severe hemorrhagic diarrhea with mortality. Helicobacter bilis and the newly recognized urease-negative organ- ism H. rodentium were isolated from microaerobic culture of feces or cecal specimens from affected mice. Dual infection with H. bilis and H. rodentium were confirmed by culture and polymerase chain reaction (PCR) in several animals. Both Helicobacter species rapidly colonized immunocompetent sentinel mice exposed to bedding from cages containing affected mice, but the sentinel remained asymptomatic. Mice with diarrhea had multifocal to segmental proliferative typhlitis, colitis, and proctitis. Several affected mice had multifocal mucosal necrosis with a few focal ulcers in the cecum, colon, and rectum. Mice with diarrhea were treated with antibiotic food wafers (1.5 mg of amoxicillin, 0.69 mg of metronidazole, and 0.185 mg of bismuth/mouse per day) previously shown to eradi- cate H. hepaticus in immunocompetent mice. Antibiotic treatment resulted in resolution of diarrhea, but not eradication of H. bilis and H. rodentium; mice continued to have positive PCR results after a 2-week treatment regimen, and clinical signs of diarrhea returned in some mice when treatment was suspended. -

Enterohepatic Lesions in SCID Mice Infected with Helicobacter Bilis
Laboratory Animal Science Vol 48, No 4 Copyright 1998 August 1998 by the American Association for Laboratory Animal Science Enterohepatic Lesions in SCID Mice Infected with Helicobacter bilis Craig L. Franklin, Lela K. Riley, Robert S. Livingston, Catherine S. Beckwith, Cynthia L. Besch-Williford, and Reuel R. Hook, Jr. Abstract _ Helicobacter bilis is a recently identified species that colonizes the intestine and liver of mice. In immunocompetent mice, infections have been associated with mild hepatitis, and in immunocompromised mice, inflammatory bowel disease has been induced by intraperitoneal inoculation of the organism. We re- port inoculation of 6-week-old C.B-17 scid/scid mice by gastric gavage with approximately 107 H. bilis colony- forming units. Groups of mice were euthanized and necropsied 12, 24, and 36 weeks after inoculation. Mild to moderate proliferative typhlitis was evident in all mice at 12 and 36 weeks after inoculation and in most mice 24 weeks after inoculation. Mild to severe chronic active hepatitis was detected in 10 of 10 male mice and 3 of 10 female mice. These results indicate that H. bilis can cause moderate to severe enterohepatic disease in immunocompromised mice. The genus Helicobacter is a rapidly expanding genus volved in lesion development. Culture of specimens from currently containing 17 named species. Members of this mice confirmed intestinal colonization with H. hepaticus. Fox genus are microaerophilic, have curved to spiral rod mor- et al. reported enteric lesions in immunocompetent germ- phology, and are motile by flagella that vary in number free Swiss Webster mice infected with H. hepaticus (15), and and location among various species (1). -

Biliary Tract Microbiota: a New Kid on the Block of Liver Diseases?
European Review for Medical and Pharmacological Sciences 2020; 24: 2750-2775 Biliary tract microbiota: a new kid on the block of liver diseases? A. NICOLETTI1, F.R. PONZIANI2, E. NARDELLA1, G. IANIRO2, A. GASBARRINI1, L. ZILERI DAL VERME2 1Internal Medicine, Gastroenterology and Hepatology, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Università Cattolica del Sacro Cuore, Rome, Italy 2Internal Medicine, Gastroenterology and Hepatology, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Rome, Italy Abstract. – The microbiome plays a crucial man body1,2. Indeed, a resident microbiota has recent- role in maintaining the homeostasis of the or- ly been described in several human environments ganism. Recent evidence has provided novel previously described as devoid of microorganisms, insights for understanding the interaction be- such as the urinary tract and the stomach3-9. Even tween the microbiota and the host. However, the 10 vast majority of such studies have analyzed the healthy placenta hosts microbial communities . interactions taking place in the intestinal tract. Bile has traditionally been considered sterile The biliary tree has traditionally been consid- under normal conditions11-14. ered sterile under normal conditions. However, The physical and chemical features of bile and the advent of metagenomic techniques has re- its antimicrobial activity were supposed to create vealed an unexpectedly rich bacterial communi- a hostile environment for bacteria. Moreover, the ty in the biliary tract. Associations between specific microbiolog- difficulty in collecting bile samples, coupled with ical patterns and inflammatory biliary diseases the lack of sensibility of culture techniques in and cancer have been recently described. Hence, detecting microbes in low-charge samples, sus- biliary dysbiosis may be a primary trigger in the tained this hypothesis for a long time. -
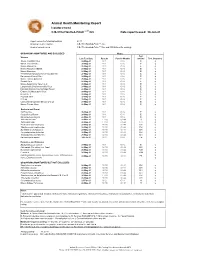
2021 ARC Health Report
Animal Health Monitoring Report Isolator reared C.B-17/IcrHanHsd-Prkdc scid /Arc Date report issued: 30-Jun-21 Report covers the following isolators: RI 27 Strains present in isolator: C.B-17/IcrHanHsd-Prkdc scid /Arc Strain of animal tested: C.B-17/IcrHanHsd-Prkdc scid /Arc and ICR Outbred for serology ORGANISMS MONITORED AND EXCLUDED Mouse Test Viruses Last Test Date Results Past 18 Months method Test frequency Mouse Hepatitis Virus 26-May-21 0 / 1 0 / 6 E q Minute Virus of Mice 26-May-21 0 / 1 0 / 6 E q Mouse Parvovirus 26-May-21 0 / 1 0 / 6 E q Murine Rotavirus (EDIM) 26-May-21 0 / 1 0 / 6 E q Mouse Norovirus 26-May-21 0 / 1 0 / 6 E q Theiler's Encephalomyelitis Virus (GD VII) 26-May-21 0 / 1 0 / 6 E q Pneumonia Virus of Mice 26-May-21 0 / 1 0 / 6 E q Murine Cytomegalovirus 26-May-21 0 / 1 0 / 6 E q Sendai Virus 26-May-21 0 / 1 0 / 6 E q Mouse Adenovirus Type 1 & 2 26-May-21 0 / 1 0 / 6 E q Lymphocytic Choriomeningitis Virus 26-May-21 0 / 1 0 / 6 E q Hantaan (Korean Haemorrhagic Fever) 26-May-21 0 / 1 0 / 6 E q Ectromelia (Mousepox) Virus 26-May-21 0 / 1 0 / 6 E q Reovirus -3 26-May-21 0 / 1 0 / 6 E q Polyoma Virus 26-May-21 0 / 1 0 / 6 E q K Virus 26-May-21 0 / 1 0 / 6 E q Lactic Dehydrogenase Elevating Virus 26-May-21 0 / 1 0 / 6 E q Mouse Thymic Virus 26-May-21 0 / 1 0 / 6 E q 00-Jan-00 Bacteria and Fungi CAR bacillus 26-May-21 0 / 1 0 / 6 E q Clostridium piliforme 26-May-21 0 / 1 0 / 6 E q Mycoplasma pulmonis 26-May-21 0 / 1 0 / 6 E q Helicobacter spp.1 26-May-21 0 / 10 0 / 44 H q Salmonella spp. -
R Graphics Output
883 | Desulfovibrio vulgaris | DvMF_2825 298701 | Desulfovibrio | DA2_3337 1121434 | Halodesulfovibrio aestuarii | AULY01000007_gene1045 207559 | Desulfovibrio alaskensis | Dde_0991 935942 | Desulfonatronum lacustre | KI912608_gene2193 159290 | Desulfonatronum | JPIK01000018_gene1259 1121448 | Desulfovibrio gigas | DGI_0655 1121445 | Desulfovibrio desulfuricans | ATUZ01000018_gene2316 525146 | Desulfovibrio desulfuricans | Ddes_0159 665942 | Desulfovibrio | HMPREF1022_02168 457398 | Desulfovibrio | HMPREF0326_00453 363253 | Lawsonia intracellularis | LI0397 882 | Desulfovibrio vulgaris | DVU_0784 1121413 | Desulfonatronovibrio hydrogenovorans | JMKT01000008_gene1463 555779 | Desulfonatronospira thiodismutans | Dthio_PD0935 690850 | Desulfovibrio africanus | Desaf_1578 643562 | Pseudodesulfovibrio aespoeensis | Daes_3115 1322246 | Pseudodesulfovibrio piezophilus | BN4_12523 641491 | Desulfovibrio desulfuricans | DND132_2573 1121440 | Desulfovibrio aminophilus | AUMA01000002_gene2198 1121456 | Desulfovibrio longus | ATVA01000018_gene290 526222 | Desulfovibrio salexigens | Desal_3460 1121451 | Desulfovibrio hydrothermalis | DESAM_21057 1121447 | Desulfovibrio frigidus | JONL01000008_gene3531 1121441 | Desulfovibrio bastinii | AUCX01000006_gene918 1121439 | Desulfovibrio alkalitolerans | dsat_0220 941449 | Desulfovibrio | dsx2_0067 1307759 | Desulfovibrio | JOMJ01000003_gene2163 1121406 | Desulfocurvus vexinensis | JAEX01000012_gene687 1304872 | Desulfovibrio magneticus | JAGC01000003_gene2904 573370 | Desulfovibrio magneticus | DMR_04750 -

Helicobacter Pylori in Thai Patients with Cholangiocarcinoma and Its Association with Biliary Inflammation and Proliferation
DOI:10.1111/j.1477-2574.2011.00423.x HPB ORIGINAL ARTICLE Helicobacter pylori in Thai patients with cholangiocarcinoma and its association with biliary inflammation and proliferation Wongwarut Boonyanugomol1,5, Chariya Chomvarin1,5, Banchob Sripa2,5, Vajarabhongsa Bhudhisawasdi3,5, Narong Khuntikeo3,5, Chariya Hahnvajanawong1,5 & Amporn Chamsuwan4 1Department of Microbiology, 2Department of Pathology, 3Department of Surgery, 4Department of Forensic Medicine and 5Liver Fluke and Cholangiocarcinoma Research Center, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand Abstracthpb_423 177..184 Objectives: To investigate whether Helicobacter spp. infection and the cagA of H. pylori are associated with hepatobiliary pathology, specifically biliary inflammation, cell proliferation and cholangiocarcinoma (CCA). Methods: Helicobacter species including H. pylori, H. bilis and H. hepaticus were detected in the speci- mens using the polymerase chain reaction (PCR). Biliary inflammation of the liver and gallbladders was semi-quantitatively graded on hematoxylin and eosin (H&E)-stained slides. Biliary proliferation was evaluated by immunohistochemistry using the Ki-67-labelling index. Results: Helicobacter pylori was found in 66.7%, 41.5% and 25.0% of the patients in the CCA, cholelithiasis and control groups (P < 0.05), respectively. By comparison, H. bilis was found in 14.9% and 9.4% of the patients with CCA and cholelithiasis, respectively (P > 0.05), and was absent in the control group. The cagA gene of H. pylori was detected in 36.2% and 9.1% of the patients with CCA and cholelithiasis, respectively (P < 0.05). Among patients with CCA, cell inflammation and proliferation in the liver and gallbladder were significantly higher among those DNA H. -

Infection with Helicobacter Pylori Induces Epithelial to Mesenchymal Transition in Human Cholangiocytes
pathogens Article Infection with Helicobacter pylori Induces Epithelial to Mesenchymal Transition in Human Cholangiocytes Prissadee Thanaphongdecha 1,2, Shannon E. Karinshak 1, Wannaporn Ittiprasert 1, Victoria H. Mann 1, Yaovalux Chamgramol 3, Chawalit Pairojkul 3, James G. Fox 4, Sutas Suttiprapa 2, Banchob Sripa 2,3,* and Paul J. Brindley 1,* 1 Research Center for Neglected Tropical Diseases of Poverty, Department of Microbiology, Immunology and Tropical Medicine, School of Medicine & Health Sciences, The George Washington University, Washington, DC 20037, USA; [email protected] (P.T.); [email protected] (S.E.K.); [email protected] (W.I.); [email protected] (V.H.M.) 2 Tropical Disease Research Laboratory, Faculty of Medicine, Khon Kaen University, Khon Kaen 40002, Thailand; [email protected] 3 Department of Pathology, Faculty of Medicine, Khon Kaen University, Khon Kaen 40002, Thailand; [email protected] (Y.C.); [email protected] (C.P.) 4 Division of Comparative Medicine, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; [email protected] * Correspondence: [email protected] (B.S.); [email protected] (P.J.B.) Received: 4 October 2020; Accepted: 18 November 2020; Published: 21 November 2020 Abstract: Recent reports suggest that the East Asian liver fluke infection, caused by Opisthorchis viverrini, which is implicated in opisthorchiasis-associated cholangiocarcinoma, serves as a reservoir of Helicobacter pylori. The opisthorchiasis-affected cholangiocytes that line the intrahepatic biliary tract are considered to be the cell of origin of this malignancy. Here, we investigated interactions in vitro among human cholangiocytes, Helicobacter pylori strain NCTC 11637, and the congeneric bacillus, Helicobacter bilis. Exposure to increasing numbers of H. -

Spiral Bacteria in the Human Stomach: the Gastric Helicobacters Andre Dubois, M.D., Ph.D
Synopses Spiral Bacteria in the Human Stomach: The Gastric Helicobacters Andre Dubois, M.D., Ph.D. Digestive Diseases Division, Department of Medicine, Uniformed Services University of the Health Sciences, Bethesda, Maryland, USA During the past decade, Helicobacter pylori has become recognized as one of the most common human pathogens, colonizing the gastric mucosa of almost all persons exposed to poor hygienic conditions from childhood. It also is often found, albeit with a lower frequency, in groups of high socioeconomic status. H. pylori causes chronic active gastritis and is a major factor in the pathogenesis of duodenal ulcers and, to a lesser extent, gastric ulcers. In addition, the presence of this bacterium is now recognized as a risk factor for gastric adenocarcinoma and lymphoma. Nevertheless, most infections appear without clinical consequences. In this second decade of intensive research, it is important to understand why H. pylori is sometimes a dangerous pathogen, and to determine how it can be eradicated in those at highest risk for severe disease. At the end of the 19th century, several types of Furthermore, in June 1994, the International spirochetes and spirilla were observed for the first Agency for Research on Cancer Working Group time in the stomach of animals (1,2). Beginning at stated , “H. pylori plays a causal role in the chain of the turn of the 20th century, similar spiral bacteria events leading to cancer,” referring to adenocarci- were found in gastrectomy specimens from patients noma and lymphoma of the stomach as well as to the with gastric cancer and peptic ulcer disease (3,4). -
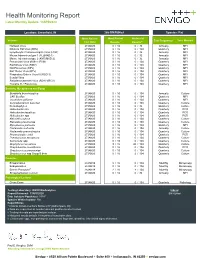
Health Monitoring Report Latest Monthly Update: 13APR2020
Health Monitoring Report Latest Monthly Update: 13APR2020 Location: Greenfield, IN 266-BN/RijHsd Species: Rat Most Recent Most Recent Historical Viruses Test Frequencyc Test Method Test Date Resultsa Resultsa,e Hantaan Virus 27JAN20 ## 0 / 16 0 / 32 Annually MFI Kilham's Rat Virus (KRV) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Lymphocytic Choriomeningitis Virus (LCM) 27JAN20 ## 0 / 16 0 / 32 Annually MFI Mouse Adenovirus type 1 (FL)(MAD-1) 27JAN20 ## 0 / 16 0 / 32 Annually MFI Mouse Adenovirus type 2 (K87)(MAD-2) 27JAN20 ## 0 / 16 0 / 32 Annually MFI Pneumonia Virus of Mice (PVM) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Rat Minute Virus (RMV) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Rat Parvovirus (RPV) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Rat Theiler Virus (RTV) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Respiratory Enteric Virus III (REO 3) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Sendai Virus 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Sialodacryoadenitis Virus (SDAV)(RCV) 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Toolan's H-1 Parvovirus 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Bacteria, Mycoplasma and Fungi Bordetella bronchiseptica 27JAN20 ## 0 / 16 0 / 104 Annually Culture CAR Bacillus 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Clostridium piliforme 27JAN20 ## 0 / 16 0 / 104 Quarterly MFI Corynebacterium kutscheri 27JAN20 ## 0 / 16 0 / 104 Quarterly Culture Dermatophytes 27JAN20 ## 0 / 16 0 / 32 Quarterly Culture Helicobacter bilis 27JAN20 ## 0 / 16 0 / 104 Quarterly PCR Helicobacter hepaticus 27JAN20 ## 0 / 16 0 / 104 Quarterly PCR Helicobacter -

Prevalence of Viral, Bacterial and Parasitological Diseases in Rats and Mice Used in Research Environments in Australasia Over a 5-Y Period
RESEARCH NOTE Prevalence of viral, bacterial and parasitological diseases in rats and mice used in research environments in Australasia over a 5-y period Elizabeth F. McInnes, BVSc, PhD, FRCPath, FIATP1, Lorna Rasmussen, BVSc, DACVP, MMEdVet2, Peony Fung, MSc1, Amanda M. Auld1, Luisana Alvarez, BSc1, David A. Lawrence1, Morgan E. Quinn, BSc(Hons)1, Tammy D. Utteridge, BVSc, PhD2, Gloria M. del Fierro, DVM, PhD2, Bianca A. Vassallo, BAnVetBioSci2 & Robert Stevenson, BSc(Hons)1 Viral, bacterial and parasitological infections in rats and mice used in biomedical research continue to occur despite improved housing and biosurveillance. The presence of disease in laboratory animals can lead to spurious results for research undertaken in universities, research institutes and the pharmaceutical industry. Here the authors report the results of serological, microbiological, parasitological and molecular tests done on mice and rats from Australasia submitted to a rodent health monitoring laboratory (Cerberus Sciences) from 2004 to 2009. In tested mice, norovirus was the most prevalent virus and ectromelia virus was the least prevalent virus. In tested rats, pneumonia virus of mice was the most prevalent virus and adenoviruses 1 and 2 were the least prevalent viruses. In mice, Helicobacter hepaticus was the most prevalent bacterium, and in rats, Proteus spp. were the most prevalent bacteria. The most common positive helminthological finding in mice and rats was the presence of all pinworms (including Aspicularis spp. and Syphacia spp.). The most common positive protozoan findings in mice and rats were Chilomastix spp. and Trichomonads. Use of improved methods for health monitoring and decreasing in importance (in a particular animal housing protects the health of rats and mice in research population in a particular country). -

WO 2012/055408 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date . 3 May 2012 (03.05.2012) WO 2012/055408 Al (51) International Patent Classification: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, CI2Q 1/68 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (21) International Application Number: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, PCT/DK20 11/000120 NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, (22) International Filing Date: RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, 27 October 201 1 (27.10.201 1) TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, 61/407,122 27 October 2010 (27.10.2010) US UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, PA 2010 70455 27 October 2010 (27.10.2010) DK RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, (71) Applicant (for all designated States except US): QUAN- LT, LU, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, TIBACT A/S [DK/DK]; Kettegards Alle 30, DK-2650 SE, SI, SK, SM, TR), OAPI (BF, BJ, CF, CG, CI, CM, Hvidovre (DK). -

Helicobacter Species
Comparative genomics analysis to differentiate metabolic and virulence gene potential in gastric versus enterohepatic Helicobacter species The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Mannion, Anthony et al. "Comparative genomics analysis to differentiate metabolic and virulence gene potential in gastric versus enterohepatic Helicobacter species." BMC Genomics 19 (November 2018): 830 © 2018 The Author(s) As Published https://doi.org/10.1186/s12864-018-5171-2 Publisher Biomed Central Ltd Version Final published version Citable link http://hdl.handle.net/1721.1/119470 Terms of Use Creative Commons Attribution Detailed Terms http://creativecommons.org/licenses/by/4.0/ Mannion et al. BMC Genomics (2018) 19:830 https://doi.org/10.1186/s12864-018-5171-2 RESEARCHARTICLE Open Access Comparative genomics analysis to differentiate metabolic and virulence gene potential in gastric versus enterohepatic Helicobacter species Anthony Mannion*, Zeli Shen and James G. Fox Abstract Background: The genus Helicobacter are gram-negative, microaerobic, flagellated, mucus-inhabiting bacteria associated with gastrointestinal inflammation and classified as gastric or enterohepatic Helicobacter species (EHS) according to host species and colonization niche. While there are over 30 official species, little is known about the physiology and pathogenic mechanisms of EHS, which account for most in the genus, as well as what genetic factors differentiate gastric versus EHS, given they inhabit different hosts and colonization niches. The objective of this study was to perform a whole-genus comparative analysis of over 100 gastric versus EHS genomes in order to identify genetic determinants that distinguish these Helicobacter species and provide insights about their evolution/ adaptation to different hosts, colonization niches, and mechanisms of virulence.