Protonix Protonix
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
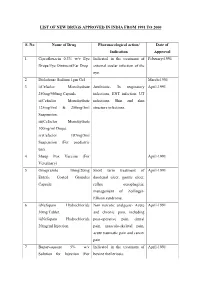
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

Proton Pump Inhibitors
MEDICATION POLICY: Proton Pump Inhibitors Generic Name: Proton Pump Inhibitors Preferred: Esomeprazole (generic), Lansoprazole (generic), Omeprazole (generic), Therapeutic Class or Brand Name: Proton Pantoprazole (generic), and Rabeprazole Pump Inhibitors (generic) Applicable Drugs (if Therapeutic Class): Non-preferred: Aciphex® (rabeprazole), Esomeprazole (generic), Lansoprazole Dexilant® (dexlansoprazole), Nexium® (generic), Omeprazole (generic), Pantoprazole (esomeprazole), Not Medically Necessary: (generic), and Rabeprazole (generic). Omeprazole/Sodium Bicarbonate (generic), Aciphex® (rabeprazole), Dexilant® Zegerid® (omeprazole/sodium bicarbonate). (dexlansoprazole), Nexium® (esomeprazole), Prevacid® (lansoprazole), Prilosec® Date of Origin: 2/1/2013 (omeprazole), and Protonix® (pantoprazole). Date Last Reviewed / Revised: 12/23/2020 Omeprazole/Sodium Bicarbonate (generic), Zegerid® (omeprazole/sodium bicarbonate). Policy also applies to any other Proton Pump Inhibitors not listed. GPI Code: 4927002000, 4927002510, 4927004000, 4927006000, 4927007010, 4927007610, 4999600260 PRIOR AUTHORIZATION CRITERIA (May be considered medically necessary when criteria I and II are met) I. Documented diagnosis of one of the following A through G: A. Gastroesophageal reflux disease (GERD). B. Erosive esophagitis. C. Gastric ulcers. D. Risk reduction of NSAID-associated gastric ulcer. E. Duodenal ulcers. F. Eradication of H. pylori. G. Hypersecretory conditions II. Non-preferred PPIs require documented trials and failures of all generic PPIs. EXCLUSION -

2018 Annual Results Presentation
2018 Annual Results Presentation Sihuan Pharmaceutical Holdings Group Ltd. 四环医药控股集团有限公司 0 Disclaimer The sole purpose of this Presentation (the “Presentation”) is to assist the recipient in deciding whether it wishes to proceed with a further investigation of Sihuan Pharmaceutical Holdings Group Ltd. (the “Company”) and it is not intended to form the basis of any decision to purchase securities, interests or assets in or of the Company. This Presentation does not constitute or contain an offer or invitation or recommendation or solicitation for the sale or purchase of securities, interests or assets in or of the Company and neither this document nor anything contained herein shall form the basis of, or be relied upon in connection with, any contract or commitment whatsoever. Any decision to purchase or subscribe for securities in any offering must be made solely on the basis of the information contained in the prospectus or offering circular issued by the company in connection with such offerings. All the information in this Presentation has been provided by the Company and has not been independently verified. No representation or warranty, express or implied, is or will be made in or in relation to, and no responsibility or liability is or will be accepted by the Company or any of its subsidiaries as to the appropriateness, accuracy, completeness or reliability of, this Presentation or any other written or oral information made available to any interested party or its advisers and any liability therefore is hereby expressly disclaimed. And no reliance should be placed on the accuracy, fairness, completeness or correctness of the information contained in this Presentation. -

PHRP March 2015
March 2015; Vol. 25(2):e2521518 doi: http://dx.doi.org/10.17061/phrp2521518 www.phrp.com.au Research Manual versus automated coding of free-text self-reported medication data in the 45 and Up Study: a validation study Danijela Gnjidica,b,i, Sallie-Anne Pearsona,c, Sarah N Hilmerb,d, Jim Basilakise, Andrea L Schaffera, Fiona M Blythb,f,g and Emily Banksg,h, on behalf of the High Risk Prescribing Investigators a Faculty of Pharmacy, University of Sydney, NSW, Australia b Sydney Medical School, University of Sydney, NSW, Australia c Sydney School of Public Health, University of Sydney, NSW, Australia d Royal North Shore Hospital and Kolling Institute of Medical Research, Sydney, NSW, Australia e School of Computing, Engineering and Mathematics, University of Western Sydney, NSW, Australia f Centre for Education and Research on Ageing (CERA), Concord Hospital, Sydney, NSW, Australia g The Sax Institute, Sydney, NSW, Australia h National Centre for Epidemiology and Population Health, Australian National University, Canberra, ACT i Corresponding author: [email protected] Article history Abstract Publication date: March 2015 Background: Increasingly, automated methods are being used to code free- Citation: Gnjidic D, Pearson S, Hilmer S, text medication data, but evidence on the validity of these methods is limited. Basilakis J, Schaffer AL, Blyth FM, Banks E. To examine the accuracy of automated coding of previously keyed Manual versus automated coding of free-text Aim: in free-text medication data compared with manual coding of original self-reported medication data in the 45 and Up Study: a validation study. Public Health handwritten free-text responses (the ‘gold standard’). -

Acid-Reducing Medications: Interactions with Harvoni and Epclusa
November 2016 | www.hepatitis.va.gov Acid-Reducing Medications: Interactions with Harvoni® and Epclusa® Sofosbuvir/Velpatasvir = Epclusa® Ledipasvir/Sofosbuvir = Harvoni® • Acid-reducing medications can decrease the effectiveness of Harvoni® and Epclusa® and could prevent successful cure. • Let your provider know if you are taking any acid-reducing medications—either prescribed to you or purchased over the counter. Types of Acid-Reducing What to Do if What to Do if Medications Taking Epclusa® Taking Harvoni® Antacids Separate taking the antacid and Separate taking the antacid and Epclusa® by 4 hours Harvoni® by 4 hours Combinations of aluminum, magnesium, and calcium. Brand names include: Alka-Seltzer® Maalox® Mylanta® Rolaids® TUMS® H2 Blockers Do not take more than the Do not take more than the recommended doses recommended doses Cimetidine (Tagamet®) Famotidine (Pepcid®) May take together with Epclusa® May take together with Harvoni® Nizatidine (Axid®) or 12 hours apart or 12 hours apart Ranitidine (Zantac®) Proton Pump Inhibitors (PPI) It is best to avoid PPIs during treat- It is best to avoid PPIs during ment with Epclusa®. If there are no treatment with Harvoni®. If there Esomeprazole (Nexium®) alternatives, follow these instructions: are no alternatives, follow these Lansoprazole (Prevacid®) instructions: Omeprazole (Prilosec®) Take Epclusa® with food Pantoprazole (Protonix®) Take the proton pump inhibitor 4 Take Harvoni® and the proton Rabeprazole (Aciphex®) hours after taking Epclusa® pump inhibitor at the same time in the morning after fasting overnight Do not take the proton pump (on an empty stomach) inhibitor at the same time as Epclusa® Do not take proton pump Do not take proton pump inhibitors inhibitors at more than the at more than the recommended dose recommended dose U.S. -

Risks of Proton Pump Inhibitors for Gastroesophageal Reflux Disease and a Diet Alternative
Journal of Nutritional Health & Food Engineering Literature Review Open Access Risks of proton pump inhibitors for gastroesophageal reflux disease and a diet alternative Abstract Volume 10 Issue 1 - 2020 Gastroesophageal reflux disease (GERD) is one of the most common digestive conditions Mark Stengler NMD treated by gastroenterologists. The most commonly prescribed drugs for GERD are proton Stengler Center for Integrative Medicine, USA pump inhibitors (PPIs) that reduce stomach acid. While these medications are effective for relieving GERD symptoms, their long-term use is associated with several side effects Correspondence: Mark Stengler NMD, Stengler Center for and chronic diseases. Several publications have questioned the long-term safety of PPIs. Integrative Medicine, 324 Encinitas Blvd. Moreover, recent studies have demonstrated that the Mediterranean Diet may be more Encinitas, CA 92024 1-760-274-2377, effective than PPIs for people suffering from GERD. Email Keywords: GERD, mediterranean diet, proton pump inhibitors, PPI, acid reflux, LES, Received: April 14, 2020 | Published: April 28, 2020 lower esophageal sphincter, omeprazole, prilosec Introduction Proton pump inhibitors to treat GERD Gastroesophageal reflux disease (GERD) is one of the most common The most commonly prescribed drugs for GERD are proton digestive conditions treated by gastroenterologists and primary care pump inhibitors (PPIs).2 PPIs currently available on prescription in doctors. Typical symptoms include heartburn, regurgitation, and the USA are: dexlansoprazole -

Proton-Pump Inhibitor (PPI) Use
Proton-Pump Inhibitor (PPI) Use The FDA has issued multiple warnings on the long-term use of PPIs. These include: increased risk of C. difficile infection1, hypomagnesemia2, and fractures of the hip, wrist, and spine3. Therefore, prudent prescribing of PPIs is warranted. The FDA recommends use of the lowest dose and shortest duration of PPI therapy appropriate for the condition being treated1-3. Patient compliance, time of administration (prior to meals), and dietary indiscretions (i.e. alcohol or irritating foods) should be assessed prior to titration of PPI doses. Indication Treatment Duration Gastroesophageal reflux disease Initial 8 week course for symptom (GERD)6 relief or esophagitis Symptomatic relief Maintenance therapy determined Omeprazole 20 mg PO by response and severity of Acute healing of erosive or ulcerative once daily disease esophagitis OR Pantoprazole 40mg PO For patients that require more Maintenance healing of erosive or once daily long-term therapy, consider a ulcerative esophagitis trial of a lower dose, on-demand therapy, or intermittent therapy to minimize exposure Stress ulcer prophylaxis Transition to PO when possible Reserve PPIs for critically ill patients Continue until resolution of with increased risk of bleeding:4,5 underlying risk factors and/or At least one of the following: critical illness - Coagulopathy (platelet count <50,000 mm3, INR >1.5, or aPTT Recommend discontinuation at >2x control) discharge, unless there is - Mechanical ventilation >48 hours Omeprazole another indication for use - History -

New and Future Drug Development for Gastroesophageal Reflux Disease
J Neurogastroenterol Motil, Vol. 20 No. 1 January, 2014 pISSN: 2093-0879 eISSN: 2093-0887 http://dx.doi.org/10.5056/jnm.2014.20.1.6 JNM Journal of Neurogastroenterology and Motility Review New and Future Drug Development for Gastroesophageal Reflux Disease Carla Maradey-Romero and Ronnie Fass* The Esophageal and Swallowing Center, Division of Gastroenterology and Hepatology, MetroHealth Medical Center, Case Western Reserve University, Cleveland, Ohio, USA Medical therapy remains the most popular treatment for gastroesophageal reflux disease (GERD). Whilst interest in drug devel- opment for GERD has declined over the last few years primarily due to the conversion of most proton pump inhibitor (PPI)’s to generic and over the counter compounds, there are still numerous areas of unmet needs in GERD. Drug development has been focused on potent histamine type 2 receptor antagonist’s, extended release PPI’s, PPI combination, potassium-competitive acid blockers, transient lower esophageal sphincter relaxation reducers, prokinetics, mucosal protectants and esophageal pain modulators. It is likely that the aforementioned compounds will be niched for specific areas of unmet need in GERD, rather than compete with the presently available anti-reflux therapies. (J Neurogastroenterol Motil 2014;20:6-16) Key Words Erosive esophagitis; Gastroesophageal reflux; Heartburn; Proton pump inhibitors Most patients with GERD fall into 1 of 3 categories: non- erosive reflux disease (NERD), erosive esophagitis (EE), and Introduction Barrett’s esophagus (BE). The 2 main phenotypes of GERD, Gastroesophageal reflux disease (GERD) is a common con- NERD and EE, appear to have different pathophysiological and dition that develops when reflux of stomach contents cause trou- clinical characteristics. -

Early Effects of Oral Administration of Omeprazole and Roxatidine on Intragastric Ph
Iida et al. / J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2012 13(1):29-34 29 Journal of Zhejiang University-SCIENCE B (Biomedicine & Biotechnology) ISSN 1673-1581 (Print); ISSN 1862-1783 (Online) www.zju.edu.cn/jzus; www.springerlink.com E-mail: [email protected] Early effects of oral administration of omeprazole and roxatidine on intragastric pH Hiroshi IIDA1, Shingo KATO1, Yusuke SEKINO1, Eiji SAKAI1, Takashi UCHIYAMA1, Hiroki ENDO1, Kunihiro HOSONO1, Yasunari SAKAMOTO1, Koji FUJITA1, Masato YONEDA1, Tomoko KOIDE1, Hirokazu TAKAHASHI1, Chikako TOKORO1, Ayumu GOTO1, Yasunobu ABE1, Noritoshi KOBAYASHI1, Kensuke KUBOTA1, Eiji GOTOH2, Shin MAEDA1, Atsushi NAKAJIMA1, Masahiko INAMORI†‡1,3 (1Gastroenterology Division, Yokohama City University School of Medicine, Yokohama 236-0004, Japan) (2Department of Medical Education, Yokohama City University School of Medicine, Yokohama 236-0004, Japan) (3Office of Postgraduate Medical Education, Yokohama City University Hospital, Yokohama 236-0004, Japan) †E-mail: [email protected] Received Mar. 14, 2011; Revision accepted Aug. 30, 2011; Crosschecked Dec. 2, 2011 Abstract: Objective: The ideal medication for the treatment of acid-related diseases, e.g., peptic ulcers, stress- related gastric bleeding, functional dyspepsia, and gastroesophageal reflux disease, should have a rapid onset of action to promote hemostasis and relieve the symptoms. The aim of our study was to investigate the inhibitory effects on gastric acid secretion of a single oral administration of a proton pump inhibitor, omeprazole 20 mg, and an H2-receptor antagonist, roxatidine 75 mg. Methods: Ten Helicobacter pylori-negative male subjects participated in this randomized, two-way crossover study. Intragastric pH was monitored continuously for 6 h after single oral admini- stration of omeprazole 20 mg and roxatidine 75 mg. -

Additional Restrictions for Select Ppis and Auto-Substitutions for Most
CALGARY ZONE Long Term Care Formulary RS - 04 SECTION SUBJECT PAGE RESTRICTED USE Proton Pump Inhibitors (PPIs) 1 of 3 pantoprazole magnesium 40 mg EC tablet rabeprazole 10 mg EC tablet Additional Restrictions for use (refer to page 3) omeprazole 20 mg DR capsule* lansoprazole 15 mg DR capsule* lansoprazole 15 mg & 30 mg oral disintegrating tablet** _____________ YY MM DD Original 98 11 25 Last update 17 03 31 Additional Restrictions for Select PPIs and Auto-substitutions For most patients, there are no clinically important differences between equivalent dosed PPIs in the treatment of most acid-related gastrointestinal conditions (1), therefor, the LTC Formulary lists only select PPIs as Formulary listings, and lists others with additional restrictions (see Table 1). The following criteria were considered in selecting the formulary listing: a lower cost, alignment with AHS formulary, and ability to meet the administration needs of most residents. An auto-substitution (therapeutic interchange) policy for oral PPI therapy was approved by the Calgary Zone LTC Pharmacy and Therapeutics committee in 1998, and continues to be in place. For residents who are unable to use the selected PPIs, a non-formulary request for funding for an alternate medication may be submitted for consideration. Refer to Table 1 for the current preferred PPI, Table 2 for comparable dosing for Auto-substitution, and to the following link Auto-substitution List - PPI-ASL-05 for the current PPI conversion table. Table 1: PPI Listings Regular Oral Use (Formulary) PPI Listing -
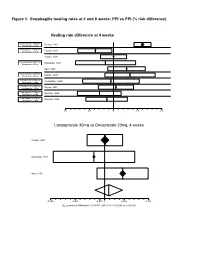
Figure 1. Esophagitis Healing Rates at 4 and 8 Weeks: PPI Vs PPI (% Risk Difference)
Figure 1. Esophagitis healing rates at 4 and 8 weeks: PPI vs PPI (% risk difference) Healing rate difference at 4 weeks Esomeprazole 40mg vs Richter, 2001 Omeprazole 20mg Lansoprazole 15mg vs Castell, 1996 Omeprazole 20mg Castell, 1996 Lansoprazole 30mg vs Hatlebakk, 1993 Omeprazole 20mg Mee, 1996 Lansoprazole 30mg vs Mulder, 1996 Omeprazole 40mg Pantoprazole 40mg vs Corinaldesi, 1995 Omeprazole 20mg Pantoprazole 40mg vs Dupas, 2001 Lansoprazole 30mg Rabeprazole 10mg vs Delchier, 2000 Omeprazole 20mg Rabeprazole 20mg vs Delchier, 2000 Omeprazole 20mg -0.2 -0.1 0 0.1 0.2 Lansoprazole 30mg vs Omeprazole 20mg, 4 weeks Castell, 1996 Hatlebakk, 1993 Mee, 1996 -0.166 -0.091 -0.016 0 0.059 0.134 DL pooled risk difference = 0.011701 (95% CI = -0.030242 to 0.053643) Figure 1 (continued) Esophagitis Healing at 4 Weeks Study Risk difference (%) (95% CI) esomeprazole 40mg vs omeprazole 20mg once daily Richter, 2001 12.0 (8.5, 15.6) lansoprazole 15mg vs omeprazole 20mg once daily Castell, 1996 -7.6 (-14.6, -0.5) lansoprazole 30mg vs omeprazole 20mg once daily Castell, 1996 0.00 (-5.4, 5.4) Hatlebakk, 1993 -3.4(-15.9, 19.1) Mee, 1996 5.4 (-2.4, 13.2) Pooled risk difference = 1.17 (95% CI -3.02, 5.36) lansoprazole 30mg vs omeprazole 40mg once daily Mulder, 1996 6.8 (-3.4, 17.0) pantoprazole 40mg vs omeprazole 20mg Corinaldesi, 1995 -1.1 (-12.9, 10.7) pantoprazole 40mg vs lansoprazole 30mg Dupas, 2001 1.0% (-6.3, 8.2) rabeprazole 10mg vs omeprazole 20mg Delchier, 2000 -5.8 (-14.6, 2.9) rabeprazole 20mg vs omeprazole 20mg Delchier, 2000 -2.8 (-11.0, 5.4) -

Package Leaflet: Information for the User Pantoprazole 20 Mg Gastro
Package leaflet: Information for the user Pantoprazole 20 mg gastro-resistant tablets Pantoprazole Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. - Keep this leaflet. You may need to read it again. - If you have any further questions, ask your doctor, pharmacist or nurse. - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. - If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Pantoprazole is and what it is used for 2. What you need to know before you take Pantoprazole 3. How to take Pantoprazole 4. Possible side effects 5. How to store Pantoprazole 6. Contents of the pack and other information 1. What Pantoprazole is and what it is used for Pantoprazole contains the active substance pantoprazole. Pantoprazole is a selective “proton pump inhibitor”, a medicine which reduces the amount of acid produced in your stomach. It is used for treating acid-related diseases of the stomach and intestine. Pantoprazole is used to treat adults and adolescents 12 years of age and above for • Symptoms (e.g. heartburn, acid regurgitation, pain on swallowing) associated to gastro-oesophageal reflux disease caused by reflux of acid from the stomach. • Long-term management of reflux oesophagitis (inflammation of the oesophagus accompanied by the regurgitation of stomach acid) and preventing its return.