Guidance for Industry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

27. Clinical Indications for Cryoprecipitate And
27. CLINICAL INDICATIONS FOR CRYOPRECIPITATE AND FIBRINOGEN CONCENTRATE Cryoprecipitate is indicated in the treatment of fibrinogen deficiency or dysfibrinogenaemia.1 Fibrinogen concentrate is licenced for the treatment of acute bleeding episodes in patients with congenital fibrinogen deficiency, including afibrinogenaemia and hypofibrinogenaemia,2 and is currently funded under the National Blood Agreement. Key messages y Fibrinogen is an essential component of the coagulation system, due to its role in initial platelet aggregation and formation of a stable fibrin clot.3 y The decision to transfuse cryoprecipitate or fibrinogen concentrate to an individual patient should take into account the relative risks and benefits.3 y The routine use of cryoprecipitate or fibrinogen concentrate is not advised in medical or critically ill patients.2,4 y Cryoprecipitate or fibrinogen concentrate may be indicated in critical bleeding if fibrinogen levels are not maintained using FFP. In the setting of major obstetric haemorrhage, early administration of cryoprecipitate or fibrinogen concentrate may be necessary.3 Clinical implications y The routine use of cryoprecipitate or fibrinogen concentrate in medical or critically ill patients with coagulopathy is not advised. The underlying causes of coagulopathy should be identified; where transfusion is considered necessary, the risks and benefits should be considered for each patient. Specialist opinion is advised for the management of disseminated intravascular coagulopathy (MED-PP18, CC-PP7).2,4 y Cryoprecipitate or fibrinogen concentrate may be indicated in critical bleeding if fibrinogen levels are not maintained using FFP. In patients with critical bleeding requiring massive transfusion, suggested doses of blood components is 3-4g (CBMT-PP10)3 in adults or as per the local Massive Transfusion Protocol. -

Hemolytic Disease of the Newborn
Intensive Care Nursery House Staff Manual Hemolytic Disease of the Newborn INTRODUCTION and DEFINITION: Hemolytic Disease of the Newborn (HDN), also known as erythroblastosis fetalis, isoimmunization, or blood group incompatibility, occurs when fetal red blood cells (RBCs), which possess an antigen that the mother lacks, cross the placenta into the maternal circulation, where they stimulate antibody production. The antibodies return to the fetal circulation and result in RBC destruction. DIFFERENTIAL DIAGNOSIS of hemolytic anemia in a newborn infant: -Isoimmunization -RBC enzyme disorders (e.g., G6PD, pyruvate kinase deficiency) -Hemoglobin synthesis disorders (e.g., alpha-thalassemias) -RBC membrane abnormalities (e.g., hereditary spherocytosis, elliptocytosis) -Hemangiomas (Kasabach Merritt syndrome) -Acquired conditions, such as sepsis, infections with TORCH or Parvovirus B19 (anemia due to RBC aplasia) and hemolysis secondary to drugs. ISOIMMUNIZATION A. Rh disease (Rh = Rhesus factor) (1) Genetics: Rh positive (+) denotes presence of D antigen. The number of antigenic sites on RBCs varies with genotype. Prevalence of genotype varies with the population. Rh negative (d/d) individuals comprise 15% of Caucasians, 5.5% of African Americans, and <1% of Asians. A sensitized Rh negative mother produces anti-Rh IgG antibodies that cross the placenta. Risk factors for antibody production include 2nd (or later) pregnancies*, maternal toxemia, paternal zygosity (D/D rather than D/d), feto-maternal compatibility in ABO system and antigen load. (2) Clinical presentation of HDN varies from mild jaundice and anemia to hydrops fetalis (with ascites, pleural and pericardial effusions). Because the placenta clears bilirubin, the chief risk to the fetus is anemia. Extramedullary hematopoiesis (due to anemia) results in hepatosplenomegaly. -

Association Between ABO and Duffy Blood Types and Circulating Chemokines and Cytokines
Genes & Immunity (2021) 22:161–171 https://doi.org/10.1038/s41435-021-00137-5 ARTICLE Association between ABO and Duffy blood types and circulating chemokines and cytokines 1 2 3 4 5 6 Sarah C. Van Alsten ● John G. Aversa ● Loredana Santo ● M. Constanza Camargo ● Troy Kemp ● Jia Liu ● 4 7 8 Wen-Yi Huang ● Joshua Sampson ● Charles S. Rabkin Received: 11 February 2021 / Revised: 30 April 2021 / Accepted: 17 May 2021 / Published online: 8 June 2021 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply 2021, corrected publication 2021 Abstract Blood group antigens are inherited traits that may play a role in immune and inflammatory processes. We investigated associations between blood groups and circulating inflammation-related molecules in 3537 non-Hispanic white participants selected from the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Whole-genome scans were used to infer blood types for 12 common antigen systems based on well-characterized single-nucleotide polymorphisms. Serum levels of 96 biomarkers were measured on multiplex fluorescent bead-based panels. We estimated marker associations with blood type using weighted linear or logistic regression models adjusted for age, sex, smoking status, and principal components of p 1234567890();,: 1234567890();,: population substructure. Bonferroni correction was used to control for multiple comparisons, with two-sided values < 0.05 considered statistically significant. Among the 1152 associations tested, 10 were statistically significant. Duffy blood type was associated with levels of CXCL6/GCP2, CXCL5/ENA78, CCL11/EOTAXIN, CXCL1/GRO, CCL2/MCP1, CCL13/ MCP4, and CCL17/TARC, whereas ABO blood type was associated with levels of sVEGFR2, sVEGFR3, and sGP130. -

Blood Product Replacement: Obstetric Hemorrhage
CMQCC OBSTETRIC HEMORRHAGE TOOLKIT Version 2.0 3/24/15 BLOOD PRODUCT REPLACEMENT: OBSTETRIC HEMORRHAGE Richard Lee, MD, Los Angeles County and University of Southern California Medical Center Laurence Shields, MD, Marian Regional Medical Center/Dignity Health Holli Mason, MD, Cedars-Sinai Medical Center Mark Rollins, MD, PhD, University of California, San Francisco Jed Gorlin, MD, Innovative Blood Resources/Memorial Blood Center, St. Paul, Minnesota Maurice Druzin, MD, Lucile Packard Children’s Hospital Stanford University Jennifer McNulty, MD, Long Beach Memorial Medical Center EXECUTIVE SUMMARY • Outcomes are improved with early and aggressive intervention. • Both emergency blood release and massive transfusion protocols should be in place. • In the setting of significant obstetric hemorrhage, resuscitation transfusion should be based on vital signs and blood loss and should not be delayed by waiting for laboratory results. • Calcium replacement will often be necessary with massive transfusion due to the citrate used for anticoagulation in blood products. • During massive transfusion resuscitation, the patient’s arterial blood gas, electrolytes, and core temperature should be monitored to guide clinical management and all transfused fluids should be warmed; direct warming of the patient should be initiated as needed to maintain euthermia and to avoid added coagulopathy. BACKGROUND AND LITERATURE REVIEW After the first several units of packed red blood cells (PRBCs) and in the face of continuing or worsening hemorrhage, aggressive transfusion therapy becomes critical. This report covers the experience with massive transfusion protocols. Lessons from military trauma units as well as civilian experience with motor vehicle accidents and massive obstetric hemorrhage have identified new principles such as earlier use of plasma (FFP/thawed plasma/plasma frozen within 24 hours/liquid plasma) and resuscitation transfusion while laboratory results are pending. -
Apheresis Donation This Quick Reference Guide Will Help You Identify the Best Donation for Your Unique Blood Type
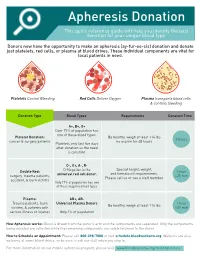
Apheresis Donation This quick reference guide will help you identify the best donation for your unique blood type. Donors now have the opportunity to make an apheresis (ay-fur-ee-sis) donation and donate just platelets, red cells, or plasma at blood drives. These individual components are vital for local patients in need. Platelets Control Bleeding Red Cells Deliver Oxygen Plasma transports blood cells & controls bleeding Donation Type Blood Types Requirements Donation Time A+, B+, O+ Over 75% of population has one of these blood types. Platelet Donation: Be healthy, weigh at least 114 lbs 2 hours cancer & surgery patients no aspirin for 48 hours Platelets only last five days after donation so the need is constant. O-, O+, A-, B- Special height, weight, Double Red: O-Negative is the 1 hour and hematocrit requirements. surgery, trauma patients, universal red cell donor. +25 min Please call us or see a staff member accident, & burn victims Only 17% of population has one of these negative blood types Plasma: AB+, AB- Trauma patients, burn Universal Plasma Donors 1 hour Be healthy, weigh at least 114 lbs victims, & patients with +30 min serious illness or injuries Only 4% of population How Apheresis works: Blood is drawn from the donor’s arm and the components are separated. Only the components being donated are collected while the remaining components are safely returned to the donor How to Schedule an Appointment: Please call 800-398-7888 or visit schedule.bloodworksnw.org. Walk-ins are also welcome at some blood drives, so be sure to ask our staff when you stop in. -

Blood Type and Transplantation & A2 Donor to B Recipient
Page 1 of 2 Blood Type and Transplantation Information for Kidney Transplant Patients Does blood type matter in transplantation? Everyone waiting for a transplant has their blood typed. You will have one of four blood types: O, A, B or AB. Your blood type is determined by the antigens that are present on your blood cells. These antigens are A or B. These antigens will be found both in your blood and on your organs. What antigen does each blood type have? Blood type O Blood type A Blood type B Blood type AB have no have A antigens. have B antigens. have both A O antigens. A B AB and B antigens. How does my body react to antigens? Your body will react to antigens that are different than your own by attacking with antibodies. Antibodies are proteins created by your immune system to attack anything that does not belong. Antibodies are the soldiers in your body’s army protecting you from foreign invasions such as viruses. Unfortunately, the antibodies cannot tell the difference between harmful viruses and beneficial transplanted organs. What blood type will my donor be? Transplants can occur between all blood types. However, when the donor’s blood type is different than yours and there are different antigens being transplanted on your new organ, your antibodies will be triggered and attack the transplanted organ. This is called rejection. Because of this, transplants usually happen between a donor and a recipient of the same blood type. This is called an identical transplant. Can I get an organ from a donor that has a different blood type than mine? Yes! If you do not have antibodies in your body against the antigens that come from the donor, your immune system should not attack the transplanted organ. -

FDA Regulation of Blood and Blood Components in the United States
FDA Regulation of Blood and Blood Components in the United States SLIDE 1 This presentation will review the FDA Regulation of Blood and Blood Components in the U.S. SLIDE 2 The FDA Center for Biologics Evaluation and Research, or CBER, Office of Blood Research and Review, called OBRR, reviews several different types of regulatory applications with respect to blood and blood components. This includes biologics license applications, called BLAs, which represent the regulatory pathway for blood components. The BLA regulatory process also applies to biological drugs such as fractionated plasma products. OBRR also regulates in-vitro diagnostic devices used for screening of collected blood, and does so also using the BLA regulatory pathway. Review of these devices as biologic licenses allows CBER to apply a higher level of manufacturing oversight, including lot release testing and pre-licensure inspection. This regulatory pathway applies to infectious disease tests for blood screening, as well as blood grouping and phenotyping reagents. SLIDE 3 The regulatory pathway for New Drug Applications, or NDAs, is most commonly used within the Center for Drug Evaluation and Research, or CDER. Within the Office of Blood, several NDA applications are reviewed each year, mostly involving solutions used for the collection of blood, such as anticoagulants and red cell nutritive solutions. Interestingly, a blood bag that does not have a solution inside is regulated as a device. If the bag has a solution, then it is a drug-device combination product, but is regulated as a drug. SLIDE 4 OBRR reviews both Class Two and Class Three devices. Class Three devices in OBRR include diagnostic tests for HIV regulated as pre-market approvals, called PMAs. -

The Johns Hopkins Comprehensive Transplant Center Incompatible Kidney Transplant Programs
The Johns Hopkins Comprehensive Transplant Center Incompatible Kidney Transplant Programs The Johns Hopkins Comprehensive Transplant Center’s Incompatible Kidney Transplantation Program allows many patients previously thought to be “incompatible” to receive the gift of life. The program is comprised of several elements: Blood Type Incompatible Kidney Transplant Program Positive Crossmatch and Sensitized Patient Program Paired Kidney Exhange Program Altruistic Donor Program Blood Type Incompatible Kidney Transplant Program More than one-third of willing live donors are turned down because their blood types are not compatible with the person to whom they wish to donate their kidney. Most of us have natural antibodies against organs from people with different blood types. These antibodies can rapidly destroy a transplanted kidney. The Blood Type Incompatible Transplant Program allows patients to receive a kidney from a live donor who has an incompatible blood type (see fig1.). Patients in this program must be willing to undergo all prescribed treatments before and after the transplant to remove harmful antibodies and decrease the risk of rejection. Figure 1: Blood Type Compatibility Chart Donor Recipient How are harmful antibodies removed? Harmful antibodies are removed with a process called plasmapheresis, a procedure similar to dialysis that removes the plasma portion of the blood where antibodies are located. The number of plasmapheresis treatments required by the recipient before surgery varies depending on the amount of harmful antibodies in their blood. After each plasmapheresis the recipient receives an intravenous infusion of immune globulin to replace antibodies needed to fight infections and help prevent harmful antibodies from returning. Once the antibodies against the donor’s blood type decrease to very low levels, the transplantation can take place. -

Recommendations for Collecting Red Blood Cells by Automated Apheresis Methods
Guidance for Industry Recommendations for Collecting Red Blood Cells by Automated Apheresis Methods Additional copies of this guidance document are available from: Office of Communication, Training and Manufacturers Assistance (HFM-40) 1401 Rockville Pike, Rockville, MD 20852-1448 (Tel) 1-800-835-4709 or 301-827-1800 (Internet) http://www.fda.gov/cber/guidelines.htm U.S. Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research (CBER) January 2001 Technical Correction February 2001 TABLE OF CONTENTS Note: Page numbering may vary for documents distributed electronically. I. INTRODUCTION ............................................................................................................. 1 II. BACKGROUND................................................................................................................ 1 III. CHANGES FROM THE DRAFT GUIDANCE .............................................................. 2 IV. RECOMMENDED DONOR SELECTION CRITERIA FOR THE AUTOMATED RED BLOOD CELL COLLECTION PROTOCOLS ..................................................... 3 V. RECOMMENDED RED BLOOD CELL PRODUCT QUALITY CONTROL............ 5 VI. REGISTRATION AND LICENSING PROCEDURES FOR THE MANUFACTURE OF RED BLOOD CELLS COLLECTED BY AUTOMATED METHODS.................. 7 VII. ADDITIONAL REQUIREMENTS.................................................................................. 9 i GUIDANCE FOR INDUSTRY Recommendations for Collecting Red Blood Cells by Automated Apheresis Methods This -

Direct Antibody Test (DAT) Positive
Direct antibody test (DAT) positive Information for patients, parents and guardians As part of your routine pregnancy screening, you However, only a small number of DAT positive have had a direct antibody test (DAT). We’ve babies will develop these problems. Babies given you this factsheet to explain what a DAT who are not DAT positive can still develop involves and what it means if your baby is DAT anaemia and jaundice. A positive DAT simply positive. If you have any further questions, tells us to look out for any signs of anaemia and please speak to a member of your healthcare jaundice. It does not necessarily mean that your team who will be pleased to advise you. baby will need treatment. What is a direct antibody test (DAT)? If we find out that you have rhesus negative During pregnancy, some of the blood between (Rh-) blood during pregnancy, we may give the mother and baby may mix. This mixing of you an injection called anti-D to stop your blood sometimes produces antibodies (proteins body making antibodies against your baby’s that are part of your body’s natural defences) blood. Occasionally this injection causes the which may become a problem for the baby. DAT result to come out positive. Babies who are DAT positive for this reason do not usually As part of routine antenatal (pregnancy) develop anaemia or jaundice. screening blood tests, your midwife will record your blood type (blood group) and check What is anaemia? whether your blood contains any antibodies Anaemia is a very common condition where the that may affect your baby’s red blood cells. -

Principles of Blood Separation and Apheresis Instrumentation
Principles of Blood Separation and Apheresis Instrumentation Dobri Kiprov, M.D., H.P. Chief, Division of Immunotherapy, California Pacific Medical Center, San Francisco, CA Medical Director, Apheresis Care Group Apheresis History Apheresis History Apheresis History Apheresis From the Greek - “to take away” Blood separation Donor apheresis Therapeutic apheresis Principles of Blood Separation Filtration Centrifugation Combined centrifugation and filtration Membrane Separation Blood is pumped through a membrane with pores allowing plasma to pass through whilst retaining blood cells. Available as a hollow fiber membrane (older devices used parallel-plate membranes) Pore diameter for plasma separation: 0.2 to 0.6μm. A number of parameters need to be closely controlled Detail of Membrane Separation Courtesy of CaridianBCT Membrane Blood Separation Trans Membrane Pressure (TMP) Too High = Hemolysis TMP Too Low = No Separation Optimal TMP = Good Separation Membrane Apheresis in the US - PrismaFlex (Gambro – Baxter) - NxStage - BBraun Filtration vs. Centrifugation Apheresis Filtration Centrifugation Minimal availability The standard in the in the USA USA • Poor industry support • Very good industry support Limited to plasma Multiple procedures (cytapheresis) exchange • Opportunity to provide • Low efficiency cellular therapies Centrifugation vs. Filtration Apheresis Centrifugation Apheresis Filtration Apheresis Blood Flow 10 – 100 ml/min 150 ml/min Efficiency of Plasma 60 – 65% 30% Removal Apheresis in Clinical Practice and Blood Banking Sickle Cell Disease Falciparum Malaria Thrombocytosis RBC WBC PLT Plasma Leukemias TTP-HUS Cell Therapies Guillain Barre Syndrome Myasthenia Gravis CIDP Autoimmune Renal Disease Hyperviscosity Syndromes Centrifugal Separation Based on the different specific gravity of the blood components. In some instruments, also based on the cellular size (Elutriation). -

Neonatal Unconjugated Hyperbilirubinemia: Risk Assessment and Management
Neonatal Unconjugated Hyperbilirubinemia: Risk Assessment and Management SHEENA BHAVSAR, PA-C PEDIATRIC FACULTY BAYLOR COLLEGE OF MEDICINE Objectives . Importance and Impact on infant health . Physiologic and Pathologic causes of hyperbilirubinemia in newborns . Guide to a systematic risk assessment . Management . Follow up . Common parent/provider questions Unconjugated Hyperbilirubinemia . Elevated levels of bilirubin in the blood, >95th percentile based to the hour specific nomogram . Severe hyperbilirubinemia is defined as > 25mg/dL . One of the primary causes of hospital readmission of neonates . Usually peaks between 3-5 days of life Bilirubin Encephalopathy - highly neurotoxic substance - can bind to brain tissue and cause neurologic dysfunction . ACUTE (hypotonia, lethargy, poor suck irritability, high pitched cry, intermittent hypertonia , seizures, fever, apnea, coma) VS . CHRONIC (movement disorders, auditory neuropathy, oculomotor dysfunction, GI concerns) KERNICTERUS Causes? INCREASED ERYTHROCYTE DECREASED BILIRUBIN BREAKDOWN CLEARANCE . Physiologic Jaundice . Physiologic Jaundice . Trauma . Breastmilk Jaundice . Blood Incompatibility . Anatomic Obstruction . Infection . Liver enzyme defect . Enzyme Deficiency . Drug Induced . Globin Synthesis Defect . Liver Disease . Membrane conditions . Metabolic INCREASED ERYTHROCYTE DECREASED BILIRUBIN BREAKDOWN CLEARANCE . Physiologic Jaundice . Physiologic Jaundice . Trauma . Breastmilk Jaundice . Blood Incompatibility . Anatomic Obstruction . Infection . Liver enzyme defect . Enzyme