Recommendations for Collecting Red Blood Cells by Automated Apheresis Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

27. Clinical Indications for Cryoprecipitate And
27. CLINICAL INDICATIONS FOR CRYOPRECIPITATE AND FIBRINOGEN CONCENTRATE Cryoprecipitate is indicated in the treatment of fibrinogen deficiency or dysfibrinogenaemia.1 Fibrinogen concentrate is licenced for the treatment of acute bleeding episodes in patients with congenital fibrinogen deficiency, including afibrinogenaemia and hypofibrinogenaemia,2 and is currently funded under the National Blood Agreement. Key messages y Fibrinogen is an essential component of the coagulation system, due to its role in initial platelet aggregation and formation of a stable fibrin clot.3 y The decision to transfuse cryoprecipitate or fibrinogen concentrate to an individual patient should take into account the relative risks and benefits.3 y The routine use of cryoprecipitate or fibrinogen concentrate is not advised in medical or critically ill patients.2,4 y Cryoprecipitate or fibrinogen concentrate may be indicated in critical bleeding if fibrinogen levels are not maintained using FFP. In the setting of major obstetric haemorrhage, early administration of cryoprecipitate or fibrinogen concentrate may be necessary.3 Clinical implications y The routine use of cryoprecipitate or fibrinogen concentrate in medical or critically ill patients with coagulopathy is not advised. The underlying causes of coagulopathy should be identified; where transfusion is considered necessary, the risks and benefits should be considered for each patient. Specialist opinion is advised for the management of disseminated intravascular coagulopathy (MED-PP18, CC-PP7).2,4 y Cryoprecipitate or fibrinogen concentrate may be indicated in critical bleeding if fibrinogen levels are not maintained using FFP. In patients with critical bleeding requiring massive transfusion, suggested doses of blood components is 3-4g (CBMT-PP10)3 in adults or as per the local Massive Transfusion Protocol. -

Blood, Lymph, and Immunity Joann Colville
Blood, Lymph, and Immunity Joann Colville CHAPTER OUTLINE .»: BLOOD Function Introduction Lymphatic Structures Plasma THE IMMUNE SYSTEM Cellular Components of Blood Function Red Blood Cells Immune Reactions Platelets Immunization: Protection Against Disease White Blood Cells THE LYMPHATICSYSTEM Lymph Formation Characteristics LEARNING OBJECTIVES • List and describe the functions of blood • List the functions of the lymphatic system • Describe the composition of blood plasma • Describe the structure and functions of the lymph nodes, • Describe the characteristics of mature erythrocytes spleen, thymus, tonsils, and GALT Describe the structure of the hemoglobin molecule and • List the functions of the immune system explain the fate of hemoglobin following intravascular and • Differentiate between specific and nonspecific immune extravascular hemolysis reactions • Give the origin of thrombocytes and describe their • Differentiate between cell-mediated and humoral immunity characteristics and functions • List the components involved in cell-mediated immunity • Listthe types of leukocytes and describe the functions of each and explain the role of each • Describe the formation of lymph fluid and its circulation List and describe the classes of immunoglobulins through the lymphatic system • Differentiate between active and passive immunity BLOOD vessels of the cardiovascular system. It has three main func- tions: transportation, regulation, and defense. INTRODUCTION Blood. Say the word, and some people cringe, others faint, Function and if you believe authors Bram Stoker, Stephen King, and 1. Blood is a transport system. Christopher Moore (all authors of vampire books), others • It carries oxygen, nutrients, and other essential com- drool. But no matter what you think about blood, animals pounds to every living cell in the body. -

The Diagnosis of Paroxysmal Nocturnal Hemoglobinuria: Role of Flow Cytometry
THE DIAGNOSIS OF PAROXYSMAL NOCTURNAL HEMOGLOBINURIA: ROLE OF FLOW CYTOMETRY Alberto Orfao Department of Medicine, Cancer Research Centre (IBMCC-CSIC/USAL), University of Salamanca, Salamanca, Spain Paroxysmal nocturnal hemoglobinuria (PNH) on one or multiple populations of peripheral blood is an acquired clonal disorder typically affecting red cells and/or leucocytes of PNH patients, this young adults, which involves the phosphatidylino- translating into a new diagnostic test. Because sitol glycan anchor biosynthesis class A gene of this flow cytometry became an essential tool (PIGA) coded in chromosome X, in virtually every in the diagnosis of PNH. In turn, it could be also case. The altered gene encodes for a defective pro- demonstrated that investigation of the presence of tein product, the pig-a enzyme which is involved GPI-deficient cells among circulating mature neu- in the early steps of the synthesis of glycosylphos- trophils and monocytes, increases the sensitivity phatidylinositol (GPI). This later molecule (GPI) of red blood cell screening because of the shorter acts as an anchor of a wide range of proteins to lifetime of both populations of leucocytes. Despite the cell surface. At present a large number of GPI- all the above, routine assessment of CD55 and anchor associated proteins have been described CD59 is also associated with several limitations which are expressed by one or multiple distinct due to distinct patterns of expression among dif- compartments of hematopoietic cells. The altered ferent cell populations, dim and heterogeneous PIGA gene leads to an altered GPI anchor which expression among normal individuals and the lack leads to defective expression of GPI-AP on the of experience in many labs as regards the normal cytoplasmic membrane, on the cell surface. -

Unintentional Platelet Removal by Plasmapheresis
Journal of Clinical Apheresis 16:55–60 (2001) Unintentional Platelet Removal by Plasmapheresis Jedidiah J. Perdue,1 Linda K. Chandler,2 Sara K. Vesely,1 Deanna S. Duvall,2 Ronald O. Gilcher,2 James W. Smith,2 and James N. George1* 1Hematology-Oncology Section, Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma 2Oklahoma Blood Institute, Oklahoma City, Oklahoma Therapeutic plasmapheresis may remove platelets as well as plasma. Unintentional platelet loss, if not recognized, may lead to inappropriate patient assessment and treatment. A patient with thrombotic thrombocytopenic purpura- hemolytic uremic syndrome (TTP-HUS) is reported in whom persistent thrombocytopenia was interpreted as continuing active disease; thrombocytopenia resolved only after plasma exchange treatments were stopped. This observation prompted a systematic study of platelet loss with plasmapheresis. Data are reported on platelet loss during 432 apheresis procedures in 71 patients with six disease categories using three different instruments. Com- paring the first procedure recorded for each patient, there was a significant difference among instrument types ,than with the COBE Spectra (1.6% (21 ס P<0.001); platelet loss was greater with the Fresenius AS 104 (17.5%, N) .With all procedures, platelet loss ranged from 0 to 71% .(24 ס or the Haemonetics LN9000 (2.6%, N (26 ס N Among disease categories, platelet loss was greater in patients with dysproteinemias who were treated for hyper- viscosity symptoms. Absolute platelet loss with the first recorded apheresis procedure, in the 34 patients who had a normal platelet count before the procedure, was also greater with the AS 104 (2.23 × 1011 platelets) than with the Spectra (0.29 × 1011 platelets) or the LN9000 (0.37 × 1011 platelets). -

Platelet-Rich Plasmapheresis: a Meta-Analysis of Clinical Outcomes and Costs
THE jOURNAL OF EXTRA-CORPOREAL TECHNOLOGY Original Article Platelet-Rich Plasmapheresis: A Meta-Analysis of Clinical Outcomes and Costs Chris Brown Mahoney , PhD Industrial Relations Center, Carlson School of Management, University of Minnesota, Minneapolis, MN Keywords: platelet-rich plasmapheresis, sequestration, cardiopulmonary bypass, outcomes, economics, meta-analysis Presented at the American Society of Extra-Corporeal Technology 35th International Conference, April 3-6, 1997, Phoenix, Arizona ABSTRACT Platelet-rich plasmapheresis (PRP) just prior to cardiopulmonary bypass (CPB) surgery is used to improve post CPB hemostasis and to minimize the risks associated with exposure to allogeneic blood and its components. Meta-analysis examines evidence ofPRP's impact on clinical outcomes by integrating the results across published research studies. Data on clinical outcomes was collected from 20 pub lished studies. These outcomes, DRG payment rates, and current national average costs were used to examine the impact of PRP on costs. This study provides evidence that the use of PRP results in improved clinical outcomes when compared to the identical control groups not receiving PRP. These improved clinical out comes result in subsequent lower costs per patient in the PRP groups. All clinical outcomes analyzed were improved: blood product usage, length of stay, intensive care stay, time to extu bation, incidence of cardiovascular accident, and incidence of reoperation. The most striking differences occur in use of all blood products, particularly packed red blood cells. This study provides an example of how initial expenditure on technology used during CPB results in overall cost savings. Estimated cost savings range from $2,505.00 to $4,209.00. -
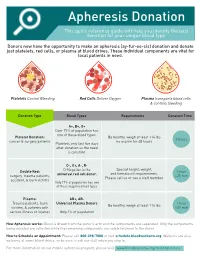
Apheresis Donation This Quick Reference Guide Will Help You Identify the Best Donation for Your Unique Blood Type
Apheresis Donation This quick reference guide will help you identify the best donation for your unique blood type. Donors now have the opportunity to make an apheresis (ay-fur-ee-sis) donation and donate just platelets, red cells, or plasma at blood drives. These individual components are vital for local patients in need. Platelets Control Bleeding Red Cells Deliver Oxygen Plasma transports blood cells & controls bleeding Donation Type Blood Types Requirements Donation Time A+, B+, O+ Over 75% of population has one of these blood types. Platelet Donation: Be healthy, weigh at least 114 lbs 2 hours cancer & surgery patients no aspirin for 48 hours Platelets only last five days after donation so the need is constant. O-, O+, A-, B- Special height, weight, Double Red: O-Negative is the 1 hour and hematocrit requirements. surgery, trauma patients, universal red cell donor. +25 min Please call us or see a staff member accident, & burn victims Only 17% of population has one of these negative blood types Plasma: AB+, AB- Trauma patients, burn Universal Plasma Donors 1 hour Be healthy, weigh at least 114 lbs victims, & patients with +30 min serious illness or injuries Only 4% of population How Apheresis works: Blood is drawn from the donor’s arm and the components are separated. Only the components being donated are collected while the remaining components are safely returned to the donor How to Schedule an Appointment: Please call 800-398-7888 or visit schedule.bloodworksnw.org. Walk-ins are also welcome at some blood drives, so be sure to ask our staff when you stop in. -

Management of Refractory Autoimmune Hemolytic Anemia Via Allogeneic Stem Cell Transplantation
Bone Marrow Transplantation (2016) 51, 1504–1506 © 2016 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 0268-3369/16 www.nature.com/bmt LETTER TO THE EDITOR Management of refractory autoimmune hemolytic anemia via allogeneic stem cell transplantation Bone Marrow Transplantation (2016) 51, 1504–1506; doi:10.1038/ urine output and nausea. Physical examination was otherwise bmt.2016.152; published online 6 June 2016 normal. Her laboratory evaluation was notable for a hematocrit of 17%, reticulocyte count of 6.8%, haptoglobin below assay limits and an Waldenström’s macroglobulinemia (WM) represents a subset LDH (lactate dehydrogenase) that was not reportable due to of lymphoplasmacytic lymphomas in which clonally related hemolysis (Table 1). Creatinine was 1.1 mg/dL, total bilirubin was lymphoplasmacytic cells secrete a monoclonal IgM Ab.1 6.7 mg/dL with a direct bilirubin of 0.3 mg/dL and lactate was Overproduced IgMs can act as cold agglutinins in WM. Upon 5 mmol/L. Over 4 h, her hematocrit decreased to 6% and her exposure to cooler temperatures in the periphery, they cause creatinine rose to 1.6 mg/dL. Given concern for acute hemolytic anemia via binding to the erythrocyte Ii-antigen group and anemia due to cold agglutinins, she was warmed and received classical complement cascade initiation.2 Treatment of seven units of warmed, packed RBC, broad-spectrum antibiotics, cold-agglutinin-mediated autoimmune hemolytic anemia (AIHA) high-dose steroids and underwent emergent plasmapheresis. She in WM typically targets the pathogenic B-cell clone1–4 or the also underwent hemodialysis for presumed pigment nephropathy. -

Blood Product Modifications: Leukofiltration, Irradiation and Washing
Blood Product Modifications: Leukofiltration, Irradiation and Washing 1. Leukocyte Reduction Definitions and Standards: o Process also known as leukoreduction, or leukofiltration o Applicable AABB Standards, 25th ed. Leukocyte-reduced RBCs At least 85% of original RBCs < 5 x 106 WBCs in 95% of units tested . Leukocyte-reduced Platelet Concentrates: At least 5.5 x 1010 platelets in 75% of units tested < 8.3 x 105 WBCs in 95% of units tested pH≥6.2 in at least 90% of units tested . Leukocyte-reduced Apheresis Platelets: At least 3.0 x 1011 platelets in 90% of units tested < 5.0 x 106 WBCs 95% of units tested pH≥6.2 in at least 90% of units tested Methods o Filter: “Fourth-generation” filters remove 99.99% WBCs o Apheresis methods: most apheresis machines have built-in leukoreduction mechanisms o Less efficient methods of reducing WBC content . Washing, deglycerolizing after thawing a frozen unit, centrifugation . These methods do not meet requirement of < 5.0 x 106 WBCs per unit of RBCs/apheresis platelets. Types of leukofiltration/leukoreduction o “Pre-storage” . Done within 24 hours of collection . May use inline filters at time of collection (apheresis) or post collection o “Pre-transfusion” leukoreduction/bedside leukoreduction . Done prior to transfusion . “Bedside” leukoreduction uses gravity-based filters at time of transfusion. Least desirable given variability in practice and absence of proficiency . Alternatively performed by transfusion service prior to issuing Benefits of leukoreduction o Prevention of alloimmunization to donor HLA antigens . Anti-HLA can mediate graft rejection and immune mediated destruction of platelets o Leukoreduced products are indicated for transplant recipients or patients who are likely platelet transfusion dependent o Prevention of febrile non-hemolytic transfusion reactions (FNHTR) . -

Chapter 06 Lecture Outline
Chapter 06 Lecture Outline See separate PowerPoint slides for all figures and tables pre- inserted into PowerPoint without notes. Copyright © 2016 McGraw-Hill Education. Permission required for reproduction or display. 1 Cardiovascular System: Blood © SPL/Science Source, (inset) © Andrew Syred/Science Source 2 Points to ponder • What type of tissue is blood and what are its components? • What is found in plasma? • Name the three formed elements in blood and their functions. • How does the structure of red blood cells relate to their function? • Describe the structure and function of each white blood cell. • What are disorders of red blood cells, white blood cells, and platelets? • What do you need to know before donating blood? • What are antigens and antibodies? • How are ABO blood types determined? • What blood types are compatible for blood transfusions? • What is the Rh factor and how is this important to pregnancy? • How does the cardiovascular system interact with other systems to maintain homeostasis? 3 6.1 Blood: An Overview What are the functions of blood? • Transportation: oxygen, nutrients, wastes, carbon dioxide, and hormones • Defense: against invasion by pathogens • Regulatory functions: body temperature, water- salt balance, and body pH 4 6.1 Blood: An Overview What is the composition of blood? • Remember: blood is a fluid connective tissue. • Formed elements are produced in red bone marrow. – Red blood cells/erythrocytes (RBCs) – White blood cells/leukocytes (WBCs) – Platelets/thrombocytes 5 6.1 Blood: An Overview What is the composition of blood? • Plasma – It consists of 91% water and 9% salts (ions) and organic molecules. – Plasma proteins are the most abundant organic molecules. -

Patient Information Leaflet – Plasma Exchange Procedure
Therapeutic Apheresis Services Patient Information Leaflet – Plasma Exchange Procedure Introduction Antibodies, which normally help to protect you from infection, can begin to attack your own This leaflet has been written to give patients healthy cells, or an over production of proteins can information about plasma exchange (sometimes cause your blood to become thicker and slow down called plasmapheresis). If you would like any more the blood flow throughout your body. A plasma information or have any questions, please ask the exchange can help improve your symptoms, doctors and nurses involved in your treatment at although this may not happen immediately. the NHS Blood and Transplant (NHSBT) Therapeutic Apheresis Services Unit. Although plasma exchange may help with symptoms, it will not normally cure your condition When you have considered the information given as it does not switch off the production of the in this leaflet, and after we have discussed the harmful antibodies or proteins. It is likely that procedure and its possible risks with you, we will this procedure will form only one part of your ask you to sign a consent form to indicate that you treatment. are happy for the procedure to go ahead. Before any further procedures we will again check that you are happy to proceed. How do we perform Plasma Exchange? What is a plasma exchange? Plasma exchange is performed using a machine Blood is made up of red cells, white cells and called a Blood Cell Separator which can separate platelets which are carried around in fluid called blood into its various parts. The machine separates plasma. -

Your Blood Cells
Page 1 of 2 Original Date The Johns Hopkins Hospital Patient Information 12/00 Oncology ReviseD/ RevieweD 6/14 Your Blood Cells Where are Blood cells are made in the bone marrow. The bone marrow blood cells is a liquid that looks like blood. It is found in several places of made? the body, such as your hips, chest bone and the middle part of your arm and leg bones. What types of • The three main types of blood cells are the red blood cells, blood cells do the white blood cells and the platelets. I have? • Red blood cells carry oxygen to all parts of the body. The normal hematocrit (or percentage of red blood cells in the blood) is 41-53%. Anemia means low red blood cells. • White blood cells fight infection. The normal white blood cell count is 4.5-11 (K/cu mm). The most important white blood cell to fight infection is the neutrophil. Forty to seventy percent (40-70%) of your white blood cells should be neutrophils. Neutropenia means your neutrophils are low, or less than 40%. • Platelets help your blood to clot and stop bleeding. The normal platelet count is 150-350 (K/cu mm). Thrombocytopenia means low platelets. How do you Your blood cells are measured by a test called the Complete measure my Blood Count (CBC) or Heme 8/Diff. You may want to keep track blood cells? of your blood counts on the back of this sheet. What When your blood counts are low, you may become anemic, get happens infections and bleed or bruise easier. -

Important Information for Female Platelet Donors
Important Information for Female Platelet Donors We are grateful for the support you provide our community blood program and especially appreciate your willingness to help save lives as a volunteer platelet donor. We also take our responsibility to provide a safe and adequate blood supply very seriously and need to share the following information regarding a change to our donor eligibility criteria for female platelet donors. We recently began performing a Human Leukocyte Antigen (HLA) antibody test on each of our current female platelet donors who have ever been pregnant. In addition, we modified our Medical History Questionnaire to ask donors whether they have been pregnant since their last donation. Platelet donors who respond yes to that question will be screened for HLA antibodies. Platelet donors will also be retested after every subsequent pregnancy. These adjustments are being made as part of our effort to reduce occurrences of Transfusion-Related Acute Lung Injury (TRALI). TRALI is a rare but serious complication of blood transfusions most commonly thought to be caused by a reaction to HLA antibodies present in the donor’s plasma. When transfused, these antibodies can sometimes cause plasma to leak into the patient’s lungs, creating fluid accumulation — a condition referred to as acute pulmonary edema. Female donors who have been pregnant are more likely than others to have these HLA antibodies in their plasma. Once the antibodies develop, they are present forever. The antibodies could be harmful if transfused into certain patients. The antibodies are present in plasma — and platelet donations contain a high volume of plasma, so our current efforts are directed at screening blood samples from female platelet donors to test for the HLA antibody.