CD59: a Long-Known Complement Inhibitor Has Advanced to a Blood Group System
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Blood, Lymph, and Immunity Joann Colville
Blood, Lymph, and Immunity Joann Colville CHAPTER OUTLINE .»: BLOOD Function Introduction Lymphatic Structures Plasma THE IMMUNE SYSTEM Cellular Components of Blood Function Red Blood Cells Immune Reactions Platelets Immunization: Protection Against Disease White Blood Cells THE LYMPHATICSYSTEM Lymph Formation Characteristics LEARNING OBJECTIVES • List and describe the functions of blood • List the functions of the lymphatic system • Describe the composition of blood plasma • Describe the structure and functions of the lymph nodes, • Describe the characteristics of mature erythrocytes spleen, thymus, tonsils, and GALT Describe the structure of the hemoglobin molecule and • List the functions of the immune system explain the fate of hemoglobin following intravascular and • Differentiate between specific and nonspecific immune extravascular hemolysis reactions • Give the origin of thrombocytes and describe their • Differentiate between cell-mediated and humoral immunity characteristics and functions • List the components involved in cell-mediated immunity • Listthe types of leukocytes and describe the functions of each and explain the role of each • Describe the formation of lymph fluid and its circulation List and describe the classes of immunoglobulins through the lymphatic system • Differentiate between active and passive immunity BLOOD vessels of the cardiovascular system. It has three main func- tions: transportation, regulation, and defense. INTRODUCTION Blood. Say the word, and some people cringe, others faint, Function and if you believe authors Bram Stoker, Stephen King, and 1. Blood is a transport system. Christopher Moore (all authors of vampire books), others • It carries oxygen, nutrients, and other essential com- drool. But no matter what you think about blood, animals pounds to every living cell in the body. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Role of C5ar1 and C5L2 Receptors in Ischemia-Reperfusion Injury
Journal of Clinical Medicine Article Role of C5aR1 and C5L2 Receptors in Ischemia-Reperfusion Injury Carlos Arias-Cabrales 1,* , Eva Rodriguez-Garcia 1, Javier Gimeno 2, David Benito 3, María José Pérez-Sáez 1, Dolores Redondo-Pachón 1, Anna Buxeda 1, Carla Burballa 1, Marta Crespo 1, Marta Riera 3,* and Julio Pascual 1,* 1 Department of Nephrology, Parc de Salut Mar, 08003 Barcelona, Spain; [email protected] (E.R.-G.); [email protected] (M.J.P.-S.); [email protected] (D.R.-P.); [email protected] (A.B.); [email protected] (C.B.); [email protected] (M.C.) 2 Department of Pathology, Parc de Salut Mar, 08003 Barcelona, Spain; [email protected] 3 Kidney Research Group, Hospital del Mar Medical Research Institute, IMIM, 08003 Barcelona, Spain; [email protected] * Correspondence: [email protected] (C.A.-C.); [email protected] (M.R.); [email protected] (J.P.) Abstract: The role of C5a receptors (C5aR1 and C5L2) in renal ischemia-reperfusion injury (IRI) is uncertain. We generated an in vitro model of hypoxia/reoxygenation with human proximal tubule epithelial cells to mimic some IRI events. C5aR1, membrane attack complex (MAC) and factor H (FH) deposits were evaluated with immunofluorescence. Quantitative polymerase chain reaction evaluated the expression of C5aR1, C5L2 genes as well as genes related to tubular injury, inflammation, and profibrotic pathways. Additionally, C5aR1 and C5L2 deposits were evaluated in kidney graft biopsies (KB) from transplant patients with delayed graft function (DGF, n = 12) and compared with Citation: Arias-Cabrales, C.; a control group (n = 8). We observed higher immunofluorescence expression of C5aR1, MAC and FH Rodriguez-Garcia, E.; Gimeno, J.; as higher expression of genes related to tubular injury, inflammatory and profibrotic pathways and Benito, D.; Pérez-Sáez, M.J.; of C5aR1 in the hypoxic cells; whereas, C5L2 gene expression was unaffected by the hypoxic stimulus. -

The Diagnosis of Paroxysmal Nocturnal Hemoglobinuria: Role of Flow Cytometry
THE DIAGNOSIS OF PAROXYSMAL NOCTURNAL HEMOGLOBINURIA: ROLE OF FLOW CYTOMETRY Alberto Orfao Department of Medicine, Cancer Research Centre (IBMCC-CSIC/USAL), University of Salamanca, Salamanca, Spain Paroxysmal nocturnal hemoglobinuria (PNH) on one or multiple populations of peripheral blood is an acquired clonal disorder typically affecting red cells and/or leucocytes of PNH patients, this young adults, which involves the phosphatidylino- translating into a new diagnostic test. Because sitol glycan anchor biosynthesis class A gene of this flow cytometry became an essential tool (PIGA) coded in chromosome X, in virtually every in the diagnosis of PNH. In turn, it could be also case. The altered gene encodes for a defective pro- demonstrated that investigation of the presence of tein product, the pig-a enzyme which is involved GPI-deficient cells among circulating mature neu- in the early steps of the synthesis of glycosylphos- trophils and monocytes, increases the sensitivity phatidylinositol (GPI). This later molecule (GPI) of red blood cell screening because of the shorter acts as an anchor of a wide range of proteins to lifetime of both populations of leucocytes. Despite the cell surface. At present a large number of GPI- all the above, routine assessment of CD55 and anchor associated proteins have been described CD59 is also associated with several limitations which are expressed by one or multiple distinct due to distinct patterns of expression among dif- compartments of hematopoietic cells. The altered ferent cell populations, dim and heterogeneous PIGA gene leads to an altered GPI anchor which expression among normal individuals and the lack leads to defective expression of GPI-AP on the of experience in many labs as regards the normal cytoplasmic membrane, on the cell surface. -

The Membrane Complement Regulatory Protein CD59 and Its Association with Rheumatoid Arthritis and Systemic Lupus Erythematosus
Current Medicine Research and Practice 9 (2019) 182e188 Contents lists available at ScienceDirect Current Medicine Research and Practice journal homepage: www.elsevier.com/locate/cmrp Review Article The membrane complement regulatory protein CD59 and its association with rheumatoid arthritis and systemic lupus erythematosus * Nibhriti Das a, Devyani Anand a, Bintili Biswas b, Deepa Kumari c, Monika Gandhi c, a Department of Biochemistry, All India Institute of Medical Sciences, New Delhi 110029, India b Department of Zoology, Ramjas College, University of Delhi, India c University School of Biotechnology, Guru Gobind Singh Indraprastha University, India article info abstract Article history: The complement cascade consisting of about 50 soluble and cell surface proteins is activated in auto- Received 8 May 2019 immune inflammatory disorders. This contributes to the pathological manifestations in these diseases. In Accepted 30 July 2019 normal health, the soluble and membrane complement regulatory proteins protect the host against Available online 5 August 2019 complement-mediated self-tissue injury by controlling the extent of complement activation within the desired limits for the host's benefit. CD59 is a membrane complement regulatory protein that inhibits the Keywords: formation of the terminal complement complex or membrane attack complex (C5b6789n) which is CD59 generated on complement activation by any of the three pathways, namely, the classical, alternative, and RA SLE the mannose-binding lectin pathway. Animal experiments and human studies have suggested impor- Pathophysiology tance of membrane complement proteins including CD59 in the pathophysiology of rheumatoid arthritis Disease marker (RA) and systemic lupus erythematosus (SLE). Here is a brief review on CD59 and its distribution, structure, functions, and association with RA and SLE starting with a brief introduction on the com- plement system, its activation, the biological functions, and relations of membrane complement regu- latory proteins, especially CD59, with RA and SLE. -

An Anticomplement Agent That Homes to the Damaged Brain and Promotes Recovery After Traumatic Brain Injury in Mice
An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice Marieta M. Rusevaa,1,2, Valeria Ramagliab,1, B. Paul Morgana, and Claire L. Harrisa,3 aInstitute of Infection and Immunity, School of Medicine, Cardiff University, Cardiff CF14 4XN, United Kingdom; and bDepartment of Genome Analysis, Academic Medical Center, Amsterdam 1105 AZ, The Netherlands Edited by Douglas T. Fearon, Cornell University, Cambridge, United Kingdom, and approved September 29, 2015 (received for review July 15, 2015) Activation of complement is a key determinant of neuropathology to rapidly and specifically inhibit MAC at sites of complement and disability after traumatic brain injury (TBI), and inhibition is activation, and test its therapeutic potential in experimental TBI. neuroprotective. However, systemic complement is essential to The construct, termed CD59-2a-CRIg, comprises CD59a linked fight infections, a critical complication of TBI. We describe a to CRIg via the murine IgG2a hinge. CD59a prevents assembly targeted complement inhibitor, comprising complement receptor of MAC in cell membranes (16), whereas CRIg binds C3b/iC3b of the Ig superfamily (CRIg) fused with complement regulator CD59a, deposited at sites of complement activation (17). The IgG2a designed to inhibit membrane attack complex (MAC) assembly at hinge promotes dimerization to increase ligand avidity. CD59- sites of C3b/iC3b deposition. CRIg and CD59a were linked via the 2a-CRIg protected in the TBI model, demonstrating that site- IgG2a hinge, yielding CD59-2a-CRIg dimer with increased iC3b/C3b targeted anti-MAC therapeutics may be effective in prevention binding avidity and MAC inhibitory activity. CD59-2a-CRIg inhibited of secondary neuropathology and improve neurologic recovery MAC formation and prevented complement-mediated lysis in vitro. -

Chapter 06 Lecture Outline
Chapter 06 Lecture Outline See separate PowerPoint slides for all figures and tables pre- inserted into PowerPoint without notes. Copyright © 2016 McGraw-Hill Education. Permission required for reproduction or display. 1 Cardiovascular System: Blood © SPL/Science Source, (inset) © Andrew Syred/Science Source 2 Points to ponder • What type of tissue is blood and what are its components? • What is found in plasma? • Name the three formed elements in blood and their functions. • How does the structure of red blood cells relate to their function? • Describe the structure and function of each white blood cell. • What are disorders of red blood cells, white blood cells, and platelets? • What do you need to know before donating blood? • What are antigens and antibodies? • How are ABO blood types determined? • What blood types are compatible for blood transfusions? • What is the Rh factor and how is this important to pregnancy? • How does the cardiovascular system interact with other systems to maintain homeostasis? 3 6.1 Blood: An Overview What are the functions of blood? • Transportation: oxygen, nutrients, wastes, carbon dioxide, and hormones • Defense: against invasion by pathogens • Regulatory functions: body temperature, water- salt balance, and body pH 4 6.1 Blood: An Overview What is the composition of blood? • Remember: blood is a fluid connective tissue. • Formed elements are produced in red bone marrow. – Red blood cells/erythrocytes (RBCs) – White blood cells/leukocytes (WBCs) – Platelets/thrombocytes 5 6.1 Blood: An Overview What is the composition of blood? • Plasma – It consists of 91% water and 9% salts (ions) and organic molecules. – Plasma proteins are the most abundant organic molecules. -

Quantitation of the Number of Molecules of Glycophorins C and D on Normal Red Blood Cells Using Radioiodinatedfab Fragments of Monoclonal Antibodies
Quantitation of the Number of Molecules of Glycophorins C and D on Normal Red Blood Cells Using RadioiodinatedFab Fragments of Monoclonal Antibodies By Jon Smythe, Brigitte Gardner, andDavid J. Anstee Two rat monoclonal antibodies (BRAC 1 and BRAC 1 1 ) cytes. Fabfragments of BRAC 1 1 and ERIC 10 gave values have been produced. BRAC 1 recognizes an epitope com- of 143,000 molecules GPC per red blood cell (RBC). Fab mon to the human erythrocyte membrane glycoproteins fragments of BRAC1 gave 225,000 molecules of GPC and glycophorin C (GPC) and glycophorin D (GPD). BRAC 11 GPD per RBC. These results indicate that GPC and GPD is specific for GPC. Fabfragments of these antibodies and together are sufficiently abundantto provide membrane at- BRlC 10, a murine monoclonal anti-GPC,were radioiodin- tachment sites for all ofthe protein 4.1 in normal RBCs. ated and used in quantitative binding assays to measure 0 1994 by The American Societyof Hematology. the number of GPC and GPD molecules on normal erythro- HE SHAPE AND deformability of the mature human (200,000)" and those reported for GPC (50,000).7 This nu- Downloaded from http://ashpublications.org/blood/article-pdf/83/6/1668/612763/1668.pdf by guest on 24 September 2021 T erythrocyte is controlled by a flexible two-dimensional merical differencehas led to the suggestion that a significant lattice of proteins, which together comprise the membrane proportion of protein 4.1 in normal erythrocyte membranes skeleton.' The major components of the skeleton are spec- must be bound to sites other than GPC and GPD.3 The trin, actin, ankyrin, and protein 4.1. -

Important Information for Female Platelet Donors
Important Information for Female Platelet Donors We are grateful for the support you provide our community blood program and especially appreciate your willingness to help save lives as a volunteer platelet donor. We also take our responsibility to provide a safe and adequate blood supply very seriously and need to share the following information regarding a change to our donor eligibility criteria for female platelet donors. We recently began performing a Human Leukocyte Antigen (HLA) antibody test on each of our current female platelet donors who have ever been pregnant. In addition, we modified our Medical History Questionnaire to ask donors whether they have been pregnant since their last donation. Platelet donors who respond yes to that question will be screened for HLA antibodies. Platelet donors will also be retested after every subsequent pregnancy. These adjustments are being made as part of our effort to reduce occurrences of Transfusion-Related Acute Lung Injury (TRALI). TRALI is a rare but serious complication of blood transfusions most commonly thought to be caused by a reaction to HLA antibodies present in the donor’s plasma. When transfused, these antibodies can sometimes cause plasma to leak into the patient’s lungs, creating fluid accumulation — a condition referred to as acute pulmonary edema. Female donors who have been pregnant are more likely than others to have these HLA antibodies in their plasma. Once the antibodies develop, they are present forever. The antibodies could be harmful if transfused into certain patients. The antibodies are present in plasma — and platelet donations contain a high volume of plasma, so our current efforts are directed at screening blood samples from female platelet donors to test for the HLA antibody. -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -
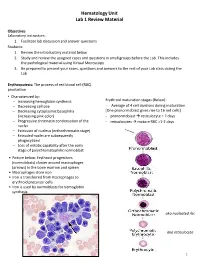
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Multiomics of Azacitidine-Treated AML Cells Reveals Variable And
Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome Kevin K. Leunga, Aaron Nguyenb, Tao Shic, Lin Tangc, Xiaochun Nid, Laure Escoubetc, Kyle J. MacBethb, Jorge DiMartinob, and James A. Wellsa,1 aDepartment of Pharmaceutical Chemistry, University of California, San Francisco, CA 94143; bEpigenetics Thematic Center of Excellence, Celgene Corporation, San Francisco, CA 94158; cDepartment of Informatics and Predictive Sciences, Celgene Corporation, San Diego, CA 92121; and dDepartment of Informatics and Predictive Sciences, Celgene Corporation, Cambridge, MA 02140 Contributed by James A. Wells, November 19, 2018 (sent for review August 23, 2018; reviewed by Rebekah Gundry, Neil L. Kelleher, and Bernd Wollscheid) Myelodysplastic syndromes (MDS) and acute myeloid leukemia of DNA methyltransferases, leading to loss of methylation in (AML) are diseases of abnormal hematopoietic differentiation newly synthesized DNA (10, 11). It was recently shown that AZA with aberrant epigenetic alterations. Azacitidine (AZA) is a DNA treatment of cervical (12, 13) and colorectal (14) cancer cells methyltransferase inhibitor widely used to treat MDS and AML, can induce interferon responses through reactivation of endoge- yet the impact of AZA on the cell-surface proteome has not been nous retroviruses. This phenomenon, termed viral mimicry, is defined. To identify potential therapeutic targets for use in com- thought to induce antitumor effects by activating and engaging bination with AZA in AML patients, we investigated the effects the immune system. of AZA treatment on four AML cell lines representing different Although AZA treatment has demonstrated clinical benefit in stages of differentiation. The effect of AZA treatment on these AML patients, additional therapeutic options are needed (8, 9).