Essential Thrombocythemia Facts No
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Updates in Mastocytosis
Updates in Mastocytosis Tryptase PD-L1 Tracy I. George, M.D. Professor of Pathology 1 Disclosure: Tracy George, M.D. Research Support / Grants None Stock/Equity (any amount) None Consulting Blueprint Medicines Novartis Employment ARUP Laboratories Speakers Bureau / Honoraria None Other None Outline • Classification • Advanced mastocytosis • A case report • Clinical trials • Other potential therapies Outline • Classification • Advanced mastocytosis • A case report • Clinical trials • Other potential therapies Mastocytosis symposium and consensus meeting on classification and diagnostic criteria for mastocytosis Boston, October 25-28, 2012 2008 WHO Classification Scheme for Myeloid Neoplasms Acute Myeloid Leukemia Chronic Myelomonocytic Leukemia Atypical Chronic Myeloid Leukemia Juvenile Myelomonocytic Leukemia Myelodysplastic Syndromes MDS/MPN, unclassifiable Chronic Myelogenous Leukemia MDS/MPN Polycythemia Vera Essential Thrombocythemia Primary Myelofibrosis Myeloproliferative Neoplasms Chronic Neutrophilic Leukemia Chronic Eosinophilic Leukemia, NOS Hypereosinophilic Syndrome Mast Cell Disease MPNs, unclassifiable Myeloid or lymphoid neoplasms Myeloid neoplasms associated with PDGFRA rearrangement associated with eosinophilia and Myeloid neoplasms associated with PDGFRB abnormalities of PDGFRA, rearrangement PDGFRB, or FGFR1 Myeloid neoplasms associated with FGFR1 rearrangement (EMS) 2017 WHO Classification Scheme for Myeloid Neoplasms Chronic Myelomonocytic Leukemia Acute Myeloid Leukemia Atypical Chronic Myeloid Leukemia Juvenile Myelomonocytic -

The Impact of Thrombophilia in the Management Of
S6 MPN III – Abstracts of the 8th International Hematology Expert Meeting / Leukemia Research 44 S1 (2016) S1–S12 Figure 1. Possible new treatment algorithm in polycythemia vera and essential thrombocythemia (CVR = cardiovascular risk factors). Adapted from: Tefferi A, Barbui T. Am J Hematol 2015;90(8):683–5 MPN III the Czech part of the international registry (“Registry”) of anagrelide (Thromboreductin®)-treated patients. The impact of thrombophilia in the management of MPN The recent analysis of the “Registry” [6] included altogether 1179 patients having MPD-T – either ET, PV or PMF according to PVSG criteria. In 812 J. Schwarz patients, the WHO/CZEMP diagnosis could be established: ET – 445 CZEMP (Czech Group for Ph– Myeloproliferative Disorders), Prague, Czech (54.8%), PMF – 206 (25.4%), PV – 107 (13.2%), and other (mostly MPN- Republic unclassifiable) – 54 (6.7%) cases. The M/F ratio was 2:3, the median age of The overall thrombotic risk in a normal healthy population is always based patients was 52 years (6–91 years) at diagnosis. The incidence of vascular on a combination of multiple risk factors present in one individual. The events was compared in the history (before entering the Registry) and risk parameters for arterial and venous events differ. MPD-T represents during follow-up (on anagrelide treatment). History and follow-up a situation in which the specific MPD-related risks (such as the JAK2 represented 4149 and 4742 patient-years, respectively. For arterial events, mutation) are combined with other risk factors present in the general there was a decrease in the incidence of events from 5.04 to 2.74 per 100 population. -

Treatment Approaches to Polycythemia Vera and Myelofibrosis
REVIEW ARTICLE Rev Hematol Mex. 2016 Apr;17(2):129-138. Treatment approaches to polycythemia vera and myelofibrosis. Palmer J, Mesa R Abstract Myeloproliferative neoplasms consist of a diverse group of disorders. Over the last 10 years, with better understanding of pathophysiology of these disorders, there are many more treatment options available to patients with these diseases. Further, improved understanding of the underlying genetic landscape has led to improved prognostication which helps identify appropriate therapeutic options. For polycythe- mia vera, initial therapy generally includes aspirin and phlebotomy. However, in patients who do not achieve an appropriate response to phlebotomy, hydroxyurea or ruxolitinib can be considered. In patients who have myelofibrosis, therapy is determined by symptom burden. In patients who have significant constitutional symptoms, a JAK inhibitor, such as ruxolitinib is an appropriate choice. There are many novel therapies under investigation for patients with myelofibrosis, including anti-fibrotic agents, novel JAK inhibitors, telomerase inhibitors and allogeneic stem cell transplant. KEYWORDS: polycythemia vera; myelofibrosis; treatment Rev Hematol Mex. 2016 abr;17(2):129-138. Enfoques terapéuticos de policitemia vera y mielofibrosis Palmer J, Mesa R Resumen Las neoplasias mieloproliferativas consisten en un diverso grupo de enfermedades. En los últimos 10 años, con mejor comprensión de estas enfermedades, hay mas opciones de tratamiento disponibles para los pacientes que las padecen. Además, el mejor entendimiento del pano- rama genético detrás de estas enfermedades ha contribuido a mejorar el pronóstico, lo que ayuda a identificar las opciones terapéuticas Mayo Clinic, Phoenix AZ, USA. adecuadas. El tratamiento inicial de la policitemia vera generalmente incluye aspirina y flebotomía. -

LEUKERAN 3 (Chlorambucil) 4 Tablets 5
1 PRESCRIBING INFORMATION ® 2 LEUKERAN 3 (chlorambucil) 4 Tablets 5 6 WARNING 7 LEUKERAN (chlorambucil) can severely suppress bone marrow function. Chlorambucil is a 8 carcinogen in humans. Chlorambucil is probably mutagenic and teratogenic in humans. 9 Chlorambucil produces human infertility (see WARNINGS and PRECAUTIONS). 10 DESCRIPTION 11 LEUKERAN (chlorambucil) was first synthesized by Everett et al. It is a bifunctional 12 alkylating agent of the nitrogen mustard type that has been found active against selected human 13 neoplastic diseases. Chlorambucil is known chemically as 4-[bis(2- 14 chlorethyl)amino]benzenebutanoic acid and has the following structural formula: 15 16 17 18 Chlorambucil hydrolyzes in water and has a pKa of 5.8. 19 LEUKERAN (chlorambucil) is available in tablet form for oral administration. Each 20 film-coated tablet contains 2 mg chlorambucil and the inactive ingredients colloidal silicon 21 dioxide, hypromellose, lactose (anhydrous), macrogol/PEG 400, microcrystalline cellulose, red 22 iron oxide, stearic acid, titanium dioxide, and yellow iron oxide. 23 CLINICAL PHARMACOLOGY 24 Chlorambucil is rapidly and completely absorbed from the gastrointestinal tract. After single 25 oral doses of 0.6 to 1.2 mg/kg, peak plasma chlorambucil levels (Cmax) are reached within 1 hour 26 and the terminal elimination half-life (t½) of the parent drug is estimated at 1.5 hours. 27 Chlorambucil undergoes rapid metabolism to phenylacetic acid mustard, the major metabolite, 28 and the combined chlorambucil and phenylacetic acid mustard urinary excretion is extremely 29 low — less than 1% in 24 hours. In a study of 12 patients given single oral doses of 0.2 mg/kg of 30 LEUKERAN, the mean dose (12 mg) adjusted (± SD) plasma chlorambucil Cmax was 31 492 ± 160 ng/mL, the AUC was 883 ± 329 ng●h/mL, t½ was 1.3 ± 0.5 hours, and the tmax was 32 0.83 ± 0.53 hours. -

Bone Marrow (Stem Cell) Transplant for Sickle Cell Disease Bone Marrow (Stem Cell) Transplant
Bone Marrow (Stem Cell) Transplant for Sickle Cell Disease Bone Marrow (Stem Cell) Transplant for Sickle Cell Disease 1 Produced by St. Jude Children’s Research Hospital Departments of Hematology, Patient Education, and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to take the place of the care and attention of your personal physician. Our goal is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specifi c treatment options should be discussed with your physician. For more general information on sickle cell disease, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital How did bone marrow (stem cell) transplants begin for children with sickle cell disease? Bone marrow (stem cell) transplants have been used for the treatment and cure of a variety of cancers, immune system diseases, and blood diseases for many years. Doctors in the United States and other countries have developed studies to treat children who have severe sickle cell disease with bone marrow (stem cell) transplants. How does a bone marrow (stem cell) transplant work? 2 In a person with sickle cell disease, the bone marrow produces red blood cells that contain hemoglobin S. This leads to the complications of sickle cell disease. • To prepare for a bone marrow (stem cell) transplant, strong medicines, called chemotherapy, are used to weaken or destroy the patient’s own bone marrow, stem cells, and infection fi ghting system. -

3Rd Year BLOOD and IMMUNOLOGY II Study Guide
BLOOD AND IMMUNOLOGY II MODULE STUDY GUIDE 3RD YEAR MBBS Contents Vision and Mission of KGMC ................................................................................................................................................................................................ Khyber Medical University: Vision ....................................................................................................................................................................................... Khyber Girls Medical College: Vision ................................................................................................................................................................................... Khyber Girls Medical College: Mission ............................................................................................................................................................................... Curriculum Committee KGMC .............................................................................................................................................................................................. Module committee ............................................................................................................................................................................................................... Outcomes of the curriculum: .............................................................................................................................................................................................. -
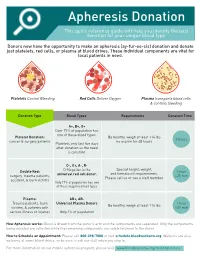
Apheresis Donation This Quick Reference Guide Will Help You Identify the Best Donation for Your Unique Blood Type
Apheresis Donation This quick reference guide will help you identify the best donation for your unique blood type. Donors now have the opportunity to make an apheresis (ay-fur-ee-sis) donation and donate just platelets, red cells, or plasma at blood drives. These individual components are vital for local patients in need. Platelets Control Bleeding Red Cells Deliver Oxygen Plasma transports blood cells & controls bleeding Donation Type Blood Types Requirements Donation Time A+, B+, O+ Over 75% of population has one of these blood types. Platelet Donation: Be healthy, weigh at least 114 lbs 2 hours cancer & surgery patients no aspirin for 48 hours Platelets only last five days after donation so the need is constant. O-, O+, A-, B- Special height, weight, Double Red: O-Negative is the 1 hour and hematocrit requirements. surgery, trauma patients, universal red cell donor. +25 min Please call us or see a staff member accident, & burn victims Only 17% of population has one of these negative blood types Plasma: AB+, AB- Trauma patients, burn Universal Plasma Donors 1 hour Be healthy, weigh at least 114 lbs victims, & patients with +30 min serious illness or injuries Only 4% of population How Apheresis works: Blood is drawn from the donor’s arm and the components are separated. Only the components being donated are collected while the remaining components are safely returned to the donor How to Schedule an Appointment: Please call 800-398-7888 or visit schedule.bloodworksnw.org. Walk-ins are also welcome at some blood drives, so be sure to ask our staff when you stop in. -

Polycythemia Vera Cancer Cluster Investigation in Northeast PA
Polycythemia Vera Cancer Cluster Investigation in Northeast PA Environmental toxic substances found historically in the PV cluster area and their potential for inducing DNA damage Investigating PV What is PV? PV is one of the diseases known as MPNs (myeloproliferative neoplasms). MPNs are a group of blood cancers where the bone marrow makes too many blood cells. Other illnesses included in this group of diseases are essential thrombocytosis (ET) and primary myelofibrosis (PMF). How does the body make blood cells? The body’s bone marrow contains billions of cells, but only a very tiny group plays a key role in forming blood cells (hematopoiesis). This group is composed of hematopoietic stem cells, which provide the body with a constant supply of all types of blood cells throughout life. What causes PV? The cause of PV is unknown. Scientists do know that a gene mutation (called the JAK2V617F mutation or “JAK2”) occurs in about 97% of PV cases. A gene mutation is a permanent change in the DNA sequence that makes up a gene inside the cells of a person’s body. The causes of the JAK2 mutation are also unknown. What is the history of the ATSDR PV investigation? • In 2005, local physicians and community members in Carbon, Luzerne, and Schuylkill counties raised concerns about the diagnosis of four cases of PV on the same rural road in the area where the three counties come together. • Residents also raised concerns about possible historical and current exposures to hazardous chemicals from various locations in the tri-county area. • The Pennsylvania Department of Health asked ATSDR for help investigating the cases and pattern of PV in this area of northeast Pennsylvania. -

Chemotherapy in Advanced Ovarian Cancer: an Overview of Randomised Clinical Trials BMJ: First Published As 10.1136/Bmj.303.6807.884 on 12 October 1991
Chemotherapy in advanced ovarian cancer: an overview of randomised clinical trials BMJ: first published as 10.1136/bmj.303.6807.884 on 12 October 1991. Downloaded from Advanced Ovarian Cancer Trialists Group Abstract cytotoxic drugs, usually doxorubicin and cyclophos- Objectives-To consider the role of platinum and phamide with or without hexamethylmelamine.45 the relative merits of single agent and combination When, however, doubt was cast on the effectiveness chemotherapy in the treatment of advanced ovarian of doxorubicin by both randomised phase III trials6-8 cancer. and phase II studies in patients not responding to Design-Formal quantitative overview using cisplatin9 several major centres adopted cisplatin plus updated individual patient data from all available cyclophosphamide as standard. Furthermore, when randomised trials (published and unpublished). other trials failed to find significant survival differences Subjects-8139 patients (6408 deaths) included in between single agent cisplatin and cisplatin in combi- 45 different trials. nation with other drugs'0 some centres reverted to Results-No firm conclusions could be reached. using single agent platinum as routine first line treat- Nevertheless, the results suggest that in terms of ment. Recently a large number oftrials have compared survival immediate platinum based treatment was cisplatin with its less nephrotoxic and neurotoxic better than non-platinum regimens (overall relative analogue carboplatin, and although follow up times risk 0-93; 95% confidence interval 0-83 to 1-05); were often short, many institutions have now adopted platinum in combination was better than single agent carboplatin as standard. platinum when used in the same dose (overall Currently it is unclear what constitutes optimal relative risk 0-85; 0*72 to 1-00); and cisplatin and chemotherapy for advanced disease and treatment carboplatin were equally effective (overall relative strategies vary both nationally and internationally. -

Myelodysplastic Syndromes Overview and Types
cancer.org | 1.800.227.2345 About Myelodysplastic Syndromes Overview and Types If you have been diagnosed with a myelodysplastic syndrome or are worried about it, you likely have a lot of questions. Learning some basics is a good place to start. ● What Are Myelodysplastic Syndromes? ● Types of Myelodysplastic Syndromes Research and Statistics See the latest estimates for new cases of myelodysplastic syndromes in the US and what research is currently being done. ● Key Statistics for Myelodysplastic Syndromes ● What's New in Myelodysplastic Syndrome Research? What Are Myelodysplastic Syndromes? Myelodysplastic syndromes (MDS) are conditions that can occur when the blood- forming cells in the bone marrow become abnormal. This leads to low numbers of one or more types of blood cells. MDS is considered a type of cancer1. Normal bone marrow 1 ____________________________________________________________________________________American Cancer Society cancer.org | 1.800.227.2345 Bone marrow is found in the middle of certain bones. It is made up of blood-forming cells, fat cells, and supporting tissues. A small fraction of the blood-forming cells are blood stem cells. Stem cells are needed to make new blood cells. There are 3 main types of blood cells: red blood cells, white blood cells, and platelets. Red blood cells pick up oxygen in the lungs and carry it to the rest of the body. These cells also bring carbon dioxide back to the lungs. Having too few red blood cells is called anemia. It can make a person feel tired and weak and look pale. Severe anemia can cause shortness of breath. White blood cells (also known as leukocytes) are important in defending the body against infection. -

Patient Information Leaflet – Plasma Exchange Procedure
Therapeutic Apheresis Services Patient Information Leaflet – Plasma Exchange Procedure Introduction Antibodies, which normally help to protect you from infection, can begin to attack your own This leaflet has been written to give patients healthy cells, or an over production of proteins can information about plasma exchange (sometimes cause your blood to become thicker and slow down called plasmapheresis). If you would like any more the blood flow throughout your body. A plasma information or have any questions, please ask the exchange can help improve your symptoms, doctors and nurses involved in your treatment at although this may not happen immediately. the NHS Blood and Transplant (NHSBT) Therapeutic Apheresis Services Unit. Although plasma exchange may help with symptoms, it will not normally cure your condition When you have considered the information given as it does not switch off the production of the in this leaflet, and after we have discussed the harmful antibodies or proteins. It is likely that procedure and its possible risks with you, we will this procedure will form only one part of your ask you to sign a consent form to indicate that you treatment. are happy for the procedure to go ahead. Before any further procedures we will again check that you are happy to proceed. How do we perform Plasma Exchange? What is a plasma exchange? Plasma exchange is performed using a machine Blood is made up of red cells, white cells and called a Blood Cell Separator which can separate platelets which are carried around in fluid called blood into its various parts. The machine separates plasma. -

B. Package Leaflet
B. PACKAGE LEAFLET 1 Package Leaflet: Information for the User Chlorambucil 2 mg tablets chlorambucil Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. - Keep this leaflet. You may need to read it again. - If you have any further questions ask your doctor or nurse. - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. - If you get any side effects, talk to your doctor or nurse. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What Chlorambucil is and what it is used for 2. What you need to know before you take Chlorambucil 3. How to take Chlorambucil 4. Possible side effects 5. How to store Chlorambucil 6. Contents of the pack and other information 1. What Chlorambucil is and what it is used for Chlorambucil contains a medicine called chlorambucil. This belongs to a group of medicines called cytotoxics (also called chemotherapy). Chlorambucil is used to treat some types of cancer and certain blood problems. It works by reducing the number of abnormal cells your body makes. Chlorambucil is used for: - Hodgkin's disease and Non-Hodgkin’s Lymphoma. Together, these form a group of diseases called lymphomas. They are cancers formed from cells of the lymphatic system. - Chronic lymphocytic leukaemia. A type of white blood cell cancer where the bone marrow produces a large number of abnormal white cells.