Circular of Information for the Use of Human Blood and Blood Components
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Guidelines for Transfusions
Guidelines for Transfu- sion Prepared by: Community Transfusion Committee Lincoln, Nebraska CHAIR: Aina Silenieks, MD MEMBERS: L. Bausch, M.D. R. Burton, M.D. S. Dunder, M.D. D. Voigt, M.D. B. J. Wilson, M.D. COMMUNITY Becky Croner REPRESENTATIVES: Ellen DiSalvo Christa Engel Phyllis Ericson Kelly Gillaspie Pat Gilles Vic Grdina Jessica Henrichs Kelly Jensen Laurel McReynolds Christina Nickel Angela Novotny Kelley Thiemann Janet Wachter Jodi Wikoff Guidelines For Transfusion Community Transfusion Committee INTRODUCTION The Community Transfusion Committee is a multidisciplinary group that meets to monitor blood utilization practices, establish guidelines for transfusion and discuss relevant transfusion related topics. It is comprised of physicians from local hospitals, invited guests, and community representatives from the hospitals’ transfusion services, nursing services, perfusion services, health information management, and the Nebraska Community Blood Bank. These Guidelines for Transfusion are reviewed and revised biannually by the Community Trans- fusion Committee to ensure that the industry’s most current practices are promoted. The Guidelines are the standard by which utilization practices are evaluated. They are also de- signed to provide helpful information to assist physicians to provide appropriate blood compo- nent therapy to patients. Appendices have been added for informational purposes and are not to be used as guidance for clinical decision making. ADULT RED CELLS A. Indications 1. One of the following a. Hypovolemia and hypoxia (signs/symptoms: syncope, dyspnea, postural hypoten- sion, tachycardia, angina, or TIA) secondary to surgery, trauma, GI tract bleeding, or intravascular hemolysis, OR b. Evidence of acute loss of 15% of total blood volume or >750 mL blood loss, OR c. -

Requirements for Blood and Blood Components Intended for Transfusion Or for Further Manufacturing Use
Requirements for Blood and Blood Components Intended for Transfusion or for Further Manufacturing Use Office of Blood Research and Review CBER, FDA Ask the FDA Session – AABB Annual Meeting October 26, 2015 1 Outline • Overview – Historical Background – Intent of the Rule – Organization of the Rule • Selected Provisions – Relevant Transfusion-Transmitted Infection – Control of Bacterial Contamination of Platelets – Medical Supervision – Donor Acknowledgement – Alternative Procedures – Donor Eligibility • Implementation 2 GAO Oversight and FDA Concerns • Publish in the form of regulations the guidelines that FDA deems essential to ensure the safety of the blood supply • Concern about the delay in requiring testing for emerging infectious agents, e.g. HTLV • Concern about blood safety and the regulations being out-of-date • Concerns about donor safety 3 Intent of the Final Rule • To better assure the safety of the blood supply and to help protect donor health • To make donor eligibility and testing requirements more consistent with current practices and to provide flexibility with regard to emerging infectious diseases • To accommodate technological advances • To establish requirements for donor education, donor history, and donor testing 4 Organization of the Final Rule - 1 • A. General • B. Definitions (§§ 606.3, 610.39, 630.3, 640.125) • C. Standard Operating Procedures (§ 606.100) • D. Control of Bacterial Contamination of Platelets (§ 606.145) • E. Records (§ 606.160) • F. Test Requirements (§§ 610.40, 640.5, 640.71(a)) • G. Donor Deferral (§ 610.41) 5 Organization of the Final Rule - 2 • H. Purpose and Scope (§ 630.1) • I. Medical Supervision (§ 630.5) • J. General Donor Eligibility Requirements(§ 630.10) • K. Donor Eligibility Requirements Specific to Whole Blood, Red Blood Cells and Plasma Collected by Apheresis (§ 630.15) • L. -

27. Clinical Indications for Cryoprecipitate And
27. CLINICAL INDICATIONS FOR CRYOPRECIPITATE AND FIBRINOGEN CONCENTRATE Cryoprecipitate is indicated in the treatment of fibrinogen deficiency or dysfibrinogenaemia.1 Fibrinogen concentrate is licenced for the treatment of acute bleeding episodes in patients with congenital fibrinogen deficiency, including afibrinogenaemia and hypofibrinogenaemia,2 and is currently funded under the National Blood Agreement. Key messages y Fibrinogen is an essential component of the coagulation system, due to its role in initial platelet aggregation and formation of a stable fibrin clot.3 y The decision to transfuse cryoprecipitate or fibrinogen concentrate to an individual patient should take into account the relative risks and benefits.3 y The routine use of cryoprecipitate or fibrinogen concentrate is not advised in medical or critically ill patients.2,4 y Cryoprecipitate or fibrinogen concentrate may be indicated in critical bleeding if fibrinogen levels are not maintained using FFP. In the setting of major obstetric haemorrhage, early administration of cryoprecipitate or fibrinogen concentrate may be necessary.3 Clinical implications y The routine use of cryoprecipitate or fibrinogen concentrate in medical or critically ill patients with coagulopathy is not advised. The underlying causes of coagulopathy should be identified; where transfusion is considered necessary, the risks and benefits should be considered for each patient. Specialist opinion is advised for the management of disseminated intravascular coagulopathy (MED-PP18, CC-PP7).2,4 y Cryoprecipitate or fibrinogen concentrate may be indicated in critical bleeding if fibrinogen levels are not maintained using FFP. In patients with critical bleeding requiring massive transfusion, suggested doses of blood components is 3-4g (CBMT-PP10)3 in adults or as per the local Massive Transfusion Protocol. -

Transfusion Service Introduction Blood/Blood Products Requests and Turnaround Time Expectations
Transfusion Service Introduction All blood products and blood components are supplied to UnityPoint Health-Meriter Hospital by the American Red Cross Blood Services. Pathology consultation is available regarding blood and/or components and dosages. ABO and Rho(D)—specific type is used whenever possible for leuko-poor packed cell transfusions. ABO-compatible blood is used for all plasma and platelet components whenever possible. For any orders involving HLA-matched components, the patient must have been HLA typed (sent to the American Red Cross) a minimum of 48 hours prior to intended infusion of the component. HLA typing is only required once. Blood components that are thawed, pooled, washed, volume-reduced, or deglycerolized for a patient will be charged to the patient even if not transfused. The charge is done because these components may not be suitable for another patient. Examples include: Autologous or directed donations Pooled cryoprecipitate 4-hour expiration Thawed fresh frozen plasma 24-hour expiration Thawed cryoprecipitate 6-hour expiration Deglycerolized red cells 24-hour expiration Washed red cells 24-hour expiration All blood components must be completely infused within 4 hours of release from UnityPoint Health-Meriter Laboratories Blood Bank, or be infused within the expiration time. Refer to UnityPoint Health -Meriter’s Blood and Blood Products Transfusion Policy #123 for additional information located on MyMeriter. Blood/Blood Products Requests and Turnaround Time Expectations Requests from UnityPoint Health-Meriter Hospital are entered in the hospital computer system and print in the UnityPoint Health- Meriter Laboratories Blood Bank. For the comfort of the patient, it is important to coordinate collection for other tests with Blood Bank specimens. -

Association Between ABO and Duffy Blood Types and Circulating Chemokines and Cytokines
Genes & Immunity (2021) 22:161–171 https://doi.org/10.1038/s41435-021-00137-5 ARTICLE Association between ABO and Duffy blood types and circulating chemokines and cytokines 1 2 3 4 5 6 Sarah C. Van Alsten ● John G. Aversa ● Loredana Santo ● M. Constanza Camargo ● Troy Kemp ● Jia Liu ● 4 7 8 Wen-Yi Huang ● Joshua Sampson ● Charles S. Rabkin Received: 11 February 2021 / Revised: 30 April 2021 / Accepted: 17 May 2021 / Published online: 8 June 2021 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply 2021, corrected publication 2021 Abstract Blood group antigens are inherited traits that may play a role in immune and inflammatory processes. We investigated associations between blood groups and circulating inflammation-related molecules in 3537 non-Hispanic white participants selected from the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Whole-genome scans were used to infer blood types for 12 common antigen systems based on well-characterized single-nucleotide polymorphisms. Serum levels of 96 biomarkers were measured on multiplex fluorescent bead-based panels. We estimated marker associations with blood type using weighted linear or logistic regression models adjusted for age, sex, smoking status, and principal components of p 1234567890();,: 1234567890();,: population substructure. Bonferroni correction was used to control for multiple comparisons, with two-sided values < 0.05 considered statistically significant. Among the 1152 associations tested, 10 were statistically significant. Duffy blood type was associated with levels of CXCL6/GCP2, CXCL5/ENA78, CCL11/EOTAXIN, CXCL1/GRO, CCL2/MCP1, CCL13/ MCP4, and CCL17/TARC, whereas ABO blood type was associated with levels of sVEGFR2, sVEGFR3, and sGP130. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -
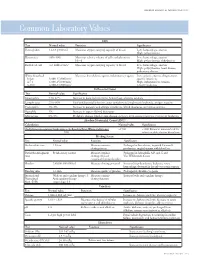
Common Laboratory Values
AmericAn AcAdemy of PediAtric dentistry Reference Manual 2006-2007 Resource Section 251 Common LaboratoryCommon Values Laboratory Values CBC Test Normal value Function Significance Hemoglobin 12-18 g/100 mL Measures oxygen carrying capacity of blood Low: hemorrhage, anemia High: polycythemia Hematocrit 35%-50% Measures relative volume of cells and plasma in Low: hemorrhage, anemia blood High: polycythemia, dehydration Red blood cell 4-6 million/mm3 Measures oxygen-carrying capacity of blood Low: hemorrhage, anemia High: polycythemia, heart disease, pulmonary disease White blood cell Measures host defense against inflammatory agents Low: aplastic anemia, drug toxicity, Infant 8,000-15,000/mm3 specific infections 4-7 y 6,000-15,000/mm3 High: inflammation, trauma, 8-18 y 4,500-13,500/mm3 toxicity, leukemia Differential Count Test Normal value Significance Neutrophils 54%-62% Increase in bacterial infections, hemorrhage, diabetic acidosis Lymphocytes 25%-30% Viral and bacterial infection, acute and chronic lymphocytic leukemia, antigen reaction Eosinophils 1%-3% Increase in parasitic and allergic conditions, blood dyscrasias, pernicious anemia Basophils 1% Increase in types of blood dyscrasias Monocytes 0%-9% Hodgkin’s disease, lipid storage disease, recovery from severe infections, monocytic leukemia Absolute Neutrophil Count (ANC) Calculation Normal value Significance (% Polymorphonuclear Leukocytes + % Bands)×Total White Cell Count >1500 <1000 Patient at increased risk for 100 infection; defer elective dental care Bleeding Screen Test -

Fresh Frozen Plasma in General Practice
Practical guide to using frozen and fresh frozen plasma in general practice Why every practice should keep a bag in the freezer Kit Sturgess; MA; VetMB; PhD; CertVR; DSAM; CertVC; FRCVS RCVS Recognised Specialist in Small Animal Medicine Advanced Practitioner in Veterinary Cardiology Introduction Keeping stock at reasonable levels in a practice is vital to good business management balancing the need to have a product available against the costs of keeping stock that is not used and potentially may go out of date and need to be replaced (as well as subtle small costs of space, stock taking, disposal of out-of-date product etc.). Practices need, therefore, to make decisions about preparedness for ‘what if’ scenarios and have protocols in place. The difficult question for many practices is how bizarre/uncommon should these scenarios be to warrant keeping a drug or treatment in stock or buying a specialist piece of equipment. This can only really be answered on an individual practice basis as it will depend on the type of cases seen as well as the likely ability of clients to want to pay if the treatment/investigation is expensive and the relationship with other local practices in terms of borrowing treatments or using equipment. It is also important to try and understand the value that a particular drug or treatment will have on morbidity and mortality of a particular condition and whether the patient could be referred onwards if necessary so not having calcium available if presented with a seizuring hypocalcaemic patient would be a significant issue in that patient’s care. -
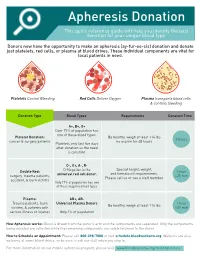
Apheresis Donation This Quick Reference Guide Will Help You Identify the Best Donation for Your Unique Blood Type
Apheresis Donation This quick reference guide will help you identify the best donation for your unique blood type. Donors now have the opportunity to make an apheresis (ay-fur-ee-sis) donation and donate just platelets, red cells, or plasma at blood drives. These individual components are vital for local patients in need. Platelets Control Bleeding Red Cells Deliver Oxygen Plasma transports blood cells & controls bleeding Donation Type Blood Types Requirements Donation Time A+, B+, O+ Over 75% of population has one of these blood types. Platelet Donation: Be healthy, weigh at least 114 lbs 2 hours cancer & surgery patients no aspirin for 48 hours Platelets only last five days after donation so the need is constant. O-, O+, A-, B- Special height, weight, Double Red: O-Negative is the 1 hour and hematocrit requirements. surgery, trauma patients, universal red cell donor. +25 min Please call us or see a staff member accident, & burn victims Only 17% of population has one of these negative blood types Plasma: AB+, AB- Trauma patients, burn Universal Plasma Donors 1 hour Be healthy, weigh at least 114 lbs victims, & patients with +30 min serious illness or injuries Only 4% of population How Apheresis works: Blood is drawn from the donor’s arm and the components are separated. Only the components being donated are collected while the remaining components are safely returned to the donor How to Schedule an Appointment: Please call 800-398-7888 or visit schedule.bloodworksnw.org. Walk-ins are also welcome at some blood drives, so be sure to ask our staff when you stop in. -

Patient's Guide to Blood Transfusions
Health Information For Patients and the Community A Patient’s Guide to Blood Transfusions Your doctor may order a blood transfusion as part of your therapy. This brochure will focus on frequently asked questions about blood products, transfusions, and the risks and benefits of the blood transfusion. PLEASE NOTE: This information is not intended to replace the medical advice of your doctor or health care provider and is intended for educational purposes only. Individual circumstances will affect your individual risks and benefits. Please discuss any questions or concerns with your doctor. What is a blood transfusion? A blood transfusion is donated blood given to patients with abnormal blood levels. The patient may have abnormal blood levels due to blood loss from trauma or surgery, or as a result of certain medical problems. The transfusion is done with one or more of the following parts of blood: red blood cells, platelets, plasma, or cryoprecipitate. What are the potential benefits of a blood transfusion? If your body does not have enough of one of the components of blood, you may develop serious life-threatening complications. • Red blood cells carry oxygen through your body to your heart and brain. Adequate oxygen is very important to maintain life. • Platelets and cryoprecipitate help to prevent or control bleeding. • Plasma replaces blood volume and also may help to prevent or control bleeding. How safe are blood transfusions? Blood donors are asked many questions about their health, behavior, and travel history in order to ensure that the blood supply is as safe as it can be. Only people who pass the survey are allowed to donate. -

Changes to the Technical Manual, 18Th Edition Monday, November 17, 2014 12:00 P.M
Changes to the Technical Manual, 18th Edition Monday, November 17, 2014 12:00 p.m. – 1:30 p.m. (ET) / 5:00p.m. – 6:30 p.m. (GMT) When this file is opened, Acrobat Reader will, by default, display the slides including the Acrobat reader controls. To return to full screen mode, hit Ctrl-L on your computer keyboard or use your mouse to click View>Full Screen on the menu bar of the Acrobat Reader program. To take the slides out of full screen mode and display the Acrobat Reader controls, simply hit the Esc key on your computer keyboard. To advance slides during the program, use the Enter, Page Down, down arrow or right arrow on your computer keyboard. To back up slides during the program, use the Page Up, up arrow of left arrow on your computer keyboard. Please remember to logon to the Live Learning Center using your email address and password to complete an evaluation of the program and speakers. At this time, advance to the next slide and wait for the audioconference to begin. A 2014 Audioconference presented to you by AABB The AABB Technical Manual 18th Edition What’s new? www.aabb.org Technical Manual by the Numbers • 1953 =year of first edition • 69 = number of authors/editors this edition • 370,378 = word count • 96 = number of methods • 60 = number of countries where TM is used www.aabb.org It’s a Process www.aabb.org Overview of Changes Major • Molecular testing • Patient blood management • Cellular therapy • Methods Minor • Throughout www.aabb.org 1: Quality Systems 2: Facilities and Safety • These 2 chapters are comprehensive discussions -

Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency
Prolonged Partial Thromboplastin Time Without Bleeding History; Fletcher Factor Deficiency Celalettin ÜSTÜN*, Anand JILLELLA*, Linda HENDRIKS*, Mary JONAH**, Ferdane KUTLAR*, Russell BURGESS*, Abdullah KUTLAR* * Section of Hematology Oncology, Department of Medicine, Medical College of Georgia, ** Department of Pathology, Medical College of Georgia, Augusta, USA ABSTRACT A 67-year-old patient was admitted to the hospital to perform an esophagogastrectomy because a lesion at the lower esophagus was strongly suspicious for cancer. Her medical history and her family his- tory were negative for bleeding tendency or thrombosis. Her activated partial thromboplastin time (aPTT) was prolonged (44 s) whereas her prothrombin time (PT) was normal (11 s) presurgery. Mixing of her plasma with normal plasma corrected her prolonged aPTT (27.9 s). Prolonged incubation shortened the patient’s aPTT (36.3 s). Fletcher factor activity was found to be 50%. The patient underwent an esopha- gogastrectomy without bleeding complications under spinal anesthesia. Fletcher factor deficiency, a ra- re disorder, should be considered in patients who have no history of bleeding tendency with a prolonged aPTT. Surgical interventions are safe in these patients. Key Words: aPTT, Surgery, Fletcher factor, Prekallikrein. Turk J Haematol 2002;19(3): 417-419 Received: 20.06.2001 Accepted: 28.07.2001 INTRODUCTION unt[2]. Hematologists are frequently consulted for preoperative coagulation abnormalities. Preoperative evaluation of hemostasis is cru- cial to assess the risk of per-and peri-operative Prolonged aPTT is not an uncommon abnor- bleeding. The most effective screening method is mality encountered during preoperative evaluati- to obtain a thorough history of bleeding[1]. on. It may indicate the presence of antiphospholi- Preoperative screening tests mostly include acti- pid antibodies or a factor deficiency in the intrinsic vated partial thromboplastin time (aPTT), proth- and/or common pathways of blood coagulation.