Transcriptome and Gene Expression Analysis of Rhynchophorus Ferrugineus (Coleoptera: Curculionidae) During Developmental Stages
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

31 First Record of Batocera Rufomaculata (De Geer, 1775) from Sunderban Biosphere Reserve, West Bengal
International Journal of Entomology Research ISSN: 2455-4758 www.entomologyjournals.com Volume 1; Issue 3; March 2016; Page No. 31-32 First record of Batocera rufomaculata (De Geer, 1775) from Sunderban biosphere reserve, West Bengal 1 Bulganin Mitra, 2 Udipta Chakraborti, 3 Olive Biswas, 4 Sankarsan Roy, 5 Kaushik Mallick, 6 Priyanka Das 1, 2, 3, 4, 6 Zoological Survey of India, Prani Vigyan Bhawan, M-Block, New Alipore, Kolkata. 5 Post Graduate Department of Zoology, Asutosh College, Kolkata Abstract Studies on Longhorn beetles (Coleoptera) in Sunderban region is very poor. Altogether, 8 species under 3 subfamilies are already reported from Sunderban Biosphere Reserve. Present communication reports Batocera rufomaculata (De Geer, 1775) for the first time from this Biosphere reserve. Keywords: Sunderban Biosphere Reserve, Cerambycidae, Lamiinae, Batocera Introduction Sunderban region in India is 9600 sq km (4200 sq km of Reserved Forest and 5400 sq km of non-forest, inhabited region) which constitutes the Sunderban Biosphere Reserve (SBR). Indian Sunderban is bound on the west by river Muriganga and on the east by rivers Harinbhahga and Raimangal. Administrative boundary of the Sunderban is spread over two districts i.e. North 24-Parganas (Hingalganj, Hasnabad, Haroa, Sandeskhali - I,II, and Minakhan blocks) and South 24-Parganas (Sagar, Namkhana, Kakdwip, Patharpratima, Kultali, Mathurapur-I,II, Jaynagar-I,II, Canning-I,II, Basanti and Gosaba blocks).The extent of mangrove Reserve Forests in Indian Sunderban is around 4260 sq km, out of which 55% is under land vegetation cover and balance 45% is under water body/ inter-tidal zone. Studies on beetles and weevils (Coleoptera) in Sunderban region is very poor. -

4 Reproductive Biology of Cerambycids
4 Reproductive Biology of Cerambycids Lawrence M. Hanks University of Illinois at Urbana-Champaign Urbana, Illinois Qiao Wang Massey University Palmerston North, New Zealand CONTENTS 4.1 Introduction .................................................................................................................................. 133 4.2 Phenology of Adults ..................................................................................................................... 134 4.3 Diet of Adults ............................................................................................................................... 138 4.4 Location of Host Plants and Mates .............................................................................................. 138 4.5 Recognition of Mates ................................................................................................................... 140 4.6 Copulation .................................................................................................................................... 141 4.7 Larval Host Plants, Oviposition Behavior, and Larval Development .......................................... 142 4.8 Mating Strategy ............................................................................................................................ 144 4.9 Conclusion .................................................................................................................................... 148 Acknowledgments ................................................................................................................................. -

Chicago Joins New York in Battle with the Asian Longhorned Beetle Therese M
Chicago Joins New York in Battle with the Asian Longhorned Beetle Therese M. Poland, Robert A. Haack, Toby R. Petrice USDA Forest Service, North Central Research Station, 1407 S. Harrison Rd., Rm. 220, E. Lansing, MI 48823 The Asian longhorned beetle, Anoplophora glabripennis (Motschulsky), was positively iden- would follow New York’s lead tified on 13 July 1998 attacking trees in an area of and that infested trees would northern Chicago known as Ravenswood. Previ- be cut, chipped, burned and ously, the only known North American occur- replaced by new trees at the rence of this Asian cerambycid beetle was in the city’s expense. Amityville area and the Brooklyn area of Long The city of Chicago ben- Island, New York, where it was discovered in efited greatly from New August 1996 (Haack et al. 1996, Cavey et al. York’s experience in imple- 1998). In New York, this woodborer has attacked menting its eradication program. With an excellent species of maple (Acer), horsechestnut (Aesculus well as 1 square mile each in Addison and in leadership team and organization, the city of hippocastanum), birch (Betula), poplar (Populus), Summit. Extensive surveys were conducted out Chicago obtained public cooperation and support willow (Salix), and elm (Ulmus) (Haack et al. to 1 ¼ miles past the outer boundary of known for the eradication program from the outset. The 1997). Because of the potential for longterm infested trees at all three locations. Survey crews media provided excellent, factual and accurate ecological and economic damage an aggressive were composed of APHIS inspectors, federal, information through extensive television, newspa- eradication program that involves locating, re- state and city employees as well as APHIS trained per, and radio coverage. -
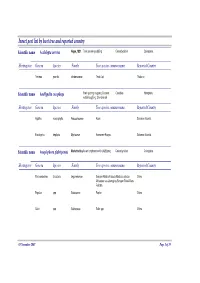
Insect Pest List by Host Tree and Reported Country
Insect pest list by host tree and reported country Scientific name Acalolepta cervina Hope, 1831 Teak canker grub|Eng Cerambycidae Coleoptera Hosting tree Genera Species Family Tree species common name Reported Country Tectona grandis Verbenaceae Teak-Jati Thailand Scientific name Amblypelta cocophaga Fruit spotting bug|eng Coconut Coreidae Hemiptera nutfall bug|Eng, Chinche del Hosting tree Genera Species Family Tree species common name Reported Country Agathis macrophylla Araucariaceae Kauri Solomon Islands Eucalyptus deglupta Myrtaceae Kamarere-Bagras Solomon Islands Scientific name Anoplophora glabripennis Motschulsky Asian longhorn beetle (ALB)|eng Cerambycidae Coleoptera Hosting tree Genera Species Family Tree species common name Reported Country Paraserianthes falcataria Leguminosae Sengon-Albizia-Falcata-Molucca albizia- China Moluccac sau-Jeungjing-Sengon-Batai-Mara- Falcata Populus spp. Salicaceae Poplar China Salix spp. Salicaceae Salix spp. China 05 November 2007 Page 1 of 35 Scientific name Aonidiella orientalis Newstead, Oriental scale|eng Diaspididae Homoptera 1894 Hosting tree Genera Species Family Tree species common name Reported Country Lovoa swynnertonii Meliaceae East African walnut Cameroon Azadirachta indica Meliaceae Melia indica-Neem Nigeria Scientific name Apethymus abdominalis Lepeletier, Tenthredinidae Hymenoptera 1823 Hosting tree Genera Species Family Tree species common name Reported Country Other Coniferous Other Coniferous Romania Scientific name Apriona germari Hope 1831 Long-horned beetle|eng Cerambycidae -

Forestry Department Food and Agriculture Organization of the United Nations
Forestry Department Food and Agriculture Organization of the United Nations Forest Health & Biosecurity Working Papers OVERVIEW OF FOREST PESTS THAILAND January 2007 Forest Resources Development Service Working Paper FBS/32E Forest Management Division FAO, Rome, Italy Forestry Department Overview of forest pests – Thailand DISCLAIMER The aim of this document is to give an overview of the forest pest1 situation in Thailand. It is not intended to be a comprehensive review. The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. © FAO 2007 1 Pest: Any species, strain or biotype of plant, animal or pathogenic agent injurious to plants or plant products (FAO, 2004). ii Overview of forest pests – Thailand TABLE OF CONTENTS Introduction..................................................................................................................... 1 Forest pests...................................................................................................................... 1 Naturally regenerating forests..................................................................................... 1 Insects ..................................................................................................................... 1 Diseases.................................................................................................................. -

09.04.2019 Several Notes Were Added
Updated: 09.04.2019 Several notes were added: #393 Viktora & Liu (2018b): Acrocyrtidus jianfeng Viktora & Liu, 2018b and Platycyrtidus yinghuii Viktora & Liu, 2018b are described from Hainan (China). Acrocyrtidus fulvus Gressitt & Rondon, 1970 is recorded from Hainan. Viktora P. & Liu B. 2018b: Notes on Asian Compsocerini Thomson, 1864 (Coleoptera, Cerambycidae, Cerambycinae) with descriptions of four new species and one new record. Folia Heyrovskyana, sries A, 26 (2): 121-142. #394 Viktora & Liu (2018c): Clytus bellus Holzschuh, 1998 (North Vietnam) is excluded from China fauna; it was included after wrong record by Hua et al (2009: 299) for Hainan. Clytus famosus Viktora & Liu, 2018c [=Clytus bellus, sensu Hua et al (2009: 299)] is described from Hainan. Demonax vendibilis Viktora & Liu, 2018c is described from Hainan. Perissus divinus Viktora & Liu, 2018c is described from Guangxi, Liuzhou County. Perissus expletus Viktora & Liu, 2018c is described from Yunnan. Perissus funicatus Viktora & Liu, 2018c is described from Yunnan. Petraphuma huangjianbini Viktora & Liu, 2018c is described from Yunnan. Petraphuma pompa Viktora & Liu, 2018c is described from Hainan. Rhaphuma caraganicola Viktora & Liu, 2018c is described from Xizang (Chayu County). Rhaphuma jianfenglingensis Viktora & Liu, 2018c is described from Hainan. Rhaphuma liyinghuii Viktora & Liu, 2018c is described from Yunnan. Xylotrechus inflexus Viktora & Liu, 2018c is described from Guangxi. Xylotrechus marketae Viktora & Liu, 2018c is described from Yunnan. Xylotrechus zhouchaoi Viktora & Liu, 2018c is described from Yunnan. Hua L.Z., Nara H., Saemulson [Samuelson] G. A. & Langafelter [Lingafelter] S. W. 2009: Iconography of Chinese Longicorn Beetles (1406 Species) in Color. Sun Yat-sen University Press, Guangzhou: 474p. Viktora P. & Liu B. -

Coleoptera: Cerambycidae) of Assam, India
Rec. zool. Surv. India: Vol. 117(1)/ 78-90, 2017 ISSN (Online) : (Applied for) DOI: 10.26515/rzsi/v117/i1/2017/117286 ISSN (Print) : 0375-1511 An updated list of cerambycid beetles (Coleoptera: Cerambycidae) of Assam, India Bulganin Mitra1*, Udipta Chakraborti1, Kaushik Mallick1, Subhrajit Bhaumik2 and Priyanka Das1 1Zoological Survey of India, Prani Vigyan Bhavan, M-Block, New Alipore, Kolkata – 700 053, West Bengal, India; [email protected] 2Post Graduate, Department of Zoology, Vidyasagar College, Kolkata – 700006, West Bengal, India Abstract consolidated updated list of cerambycid fauna of Assam and reports 95 species, 64 genera, 32 tribes and 3 subfamilies. AmongAssam isthe a threestate subfamiliesin North-East from India Assam, which subfamily is considered Lamiinae as shares a biological 49 species, hotspot. followed Present by the communication subfamily Cerambycinae is the first with 38 species and Prioninae with only 8 species. Keywords: Longhorn beetle, Assam, North-East India Introduction world, therefore this beetle family is considered as one of important coleopteran family (Agarwala & Bhattacharjee, The study on long horned beetles from the northeast 2012). This communication is the first updated Indian state Assam is very poor with many species consolidated list of cerambycid beetles from the state of awaiting discovery, study and description. Among the Assam (after complete separation from other states of NE seven sister states, cerambycid fauna of Arunachal India in 1987) which includes 95 species under 64 genera Pradesh, Tripura, Meghalaya, Manipur, Mizoram, of 32 tribes belonging to 3 subfamilies along with their Nagaland are mostly worked out by the Zoological Survey distribution. of India and some other universities and institutions. -

PRA on Apriona Species
EUROPEAN AND MEDITERRANEAN PLANT PROTECTION ORGANIZATION ORGANISATION EUROPEENNE ET MEDITERRANEENNE POUR LA PROTECTION DES PLANTES 16-22171 (13-18692) Only the yellow note is new compared to document 13-18692 Pest Risk Analysis for Apriona germari, A. japonica, A. cinerea Note: This PRA started 2011; as a result, three species of Apriona were added to the EPPO A1 List: Apriona germari, A. japonica and A. cinerea. However recent taxonomic changes have occurred with significant consequences on their geographical distributions. A. rugicollis is no longer considered as a synonym of A. germari but as a distinct species. A. japonica, which was previously considered to be a distinct species, has been synonymized with A. rugicollis. Finally, A. cinerea remains a separate species. Most of the interceptions reported in the EU as A. germari are in fact A. rugicollis. The outcomes of the PRA for these pests do not change. However A. germari has a more limited and a more tropical distribution than originally assessed, but it is considered that it could establish in Southern EPPO countries. The Panel on Phytosanitary Measures agreed with the addition of Apriona rugicollis to the A1 list. September 2013 EPPO 21 Boulevard Richard Lenoir 75011 Paris www.eppo.int [email protected] This risk assessment follows the EPPO Standard PM PM 5/3(5) Decision-support scheme for quarantine pests (available at http://archives.eppo.int/EPPOStandards/pra.htm) and uses the terminology defined in ISPM 5 Glossary of Phytosanitary Terms (available at https://www.ippc.int/index.php). This document was first elaborated by an Expert Working Group and then reviewed by the Panel on Phytosanitary Measures and if relevant other EPPO bodies. -

Core Historical Literature of Agriculture
Core Historical Literature of Agriculture Core Historical Literature of Agriculture Home Search Browse Bookbag Help The fig Table of contents | Add to bookbag First Page Page i New York State College of Agriculture At Cornell University Ithaca, N.Y. Library Front Matter Page ii Ira Judson Condit was born in 1883 at Jersey, Licking County, Ohio. After graduation from the High School at Granville in 1900 he taught in a country school at the age of 16. He received the B.Sc. degree from Ohio State University in 1905 and then spent over a year in the Division of Entomology, United States Department of Agriculture, Washington, D. C. From 1907 to 1912 he was Instructor in Botany and Horticulture at the California Polytechnic School, San Luis Obispo, where he married Caroline Callender. The following year was spent in graduate work at the University of California. In 1913 he was appointed Instructor and later Assistant Professor in the newly organised Division of Citriculture of the University, where he spec. alized in the study of subtropical fruits and published bulletins on the loquat, persimmon, avocado, carob, and caprifigs and caprification. From 1920 to 1924 he was horticulturist for the California Peach and Fig Growers zvith headquarters at Fresno and became intimately acquainted with t the fig industry and its cultural problems. Following his return to the University of California he was granted the degree of M.S. in 1928 from that institution and the Ph.D. degree from Stanford University in 1932. He has made http://chla.library.cornell.edu/cgi/t/text/text-i...dd90d19d3aa0fdc33;rgn=main;view=text;idno=3116126 (1 of 335) [1/20/2010 10:35:07 PM] Core Historical Literature of Agriculture morphological studies of the flowers of the common fig and cytological studies of over thirty species of the genus, Ficus, and for many years has investigated the problems of olive culture in California. -

60 Bionomics of Batocera Rufomaculata De Geer
Bionomics of Batocera rufomaculata De Geer (Coleoptera: Cerambycidae) in mulberry farms of Jammu and Kashmir (India) Altaf Hussain Mir Post Graduate Department of Zoology, University of Kashmir, Srinagar-190006 [email protected] Abstract: Investigations on the bionomics of Batocera rufomaculata De Geer (Coleoptera: Cerambycidae) in mulberry farms of Jammu and Kashmir state (India) demonstrated an annual life cycle with an adult emergence period of over four months from May to September; adult emergence started in May, peaked in June and ended in August. Adults required maturation feeding period of 14.00±0.561 days. Sexually mature beetles mated promiscuously and copulation period averaged 60.50±6.23 seconds. Females deposited eggs singly into the bark of host plants with an average daily and life time fecundity of 1.27 and 148.66 eggs respectively. Eggs hatched in 10.20± 1.25 days and larvae developed through 9 instars in 253.35±4.37 days. Ultimate larval instar pupated in the pupal cells, made by the ultimate larvae; adults emerged from the pupal cells after 27.5±1.35 days through circular emergence holes. Larvae as well as adults caused damage to the host plants; the latter compensated the damage caused by adults to some extent by sprouting lateral buds while as grubs caused irreparable damage and lead the host plants to death. The extent of damage caused to the host plants is also reported in the paper. [Altaf Hussain Mir. Bionomics of Batocera rufomaculata De Geer (Coleoptera: Cerambycidae) in mulberry farms of Jammu and Kashmir (India).Life Sci J 2021;18 (5):60-72]. -

Quikscan Batocera Spp Aug 2013 Def
National Plant Protection Organization, the Netherlands Quick scan number: QS. Ent.2013.05 Quick scan date : 29-08-2013 1 What is the scientific name ( if Batocera- species (longhorn beetles). Coleoptera family: Cerambycidae, Subfamily: Lamiinae, Tribus: possible up to species level + Batocerini, Genus Batocera Dejean, 1835 author, also include (sub)family and order) and English/common name The genus Batocera now has 55 recognised species, and more than 50 subspecies and varieties are known (Tavakilian of the organism? & Chevillotte, 2013; Liu et al., 2012). Recently, species of the genus Megacriodes Pascoe, 1866 have been assigned to Add picture of organism/damage if the genus Batocera (Perger & Vitali, 2012). Five species are so-called incertis sedae . Batocera species are large available and publication allowed . longhorn beetles, ranging from 30 to 85 mm ( B. hercules ) in size. For most species, only collection data are available, often without host plant records. Some species are common, others are very rare. Few species are known to cause (significant) economical damage (Sorauer, 1954; Duffy, 1968; Yang et al., 2012). Here we provide a quick scan of the possibly 12 most harmful species: species of which host records exist, that are either being intercepted in or introduced into another region of the world, and are known to cause damage. 1. Batocera davidis Deyrolle, 1878 2. Batocera gigas (Drapiez, 1819) 3. Batocera hector Thomson, 1858 - Orange spotted longhorn beetle, Dadap boktor, Dadap Longhorn Beetle 4. Batocera hercules Boisduval, 1835 5. Batocera horsfieldi (Hope, 1839) - Walnut longhorn beetle 6. Batocera lineolata Chevrolat, 1852 - White striped longhorn beetle, Populus longicorn beetle 7. -

8 March 2013, 381 P
See discussions, stats, and author profiles for this publication at: http://www.researchgate.net/publication/273257107 Mason, P. G., D. R. Gillespie & C. Vincent (Eds.) 2013. Proceedings of the Fourth International Symposium on Biological Control of Arthropods. Pucón, Chile, 4-8 March 2013, 381 p. CONFERENCE PAPER · MARCH 2013 DOWNLOADS VIEWS 626 123 3 AUTHORS, INCLUDING: Peter Mason Charles Vincent Agriculture and Agri-Food Canada Agriculture and Agri-Food Canada 96 PUBLICATIONS 738 CITATIONS 239 PUBLICATIONS 1,902 CITATIONS SEE PROFILE SEE PROFILE Available from: Charles Vincent Retrieved on: 13 August 2015 The correct citation of this work is: Peter G. Mason, David R. Gillespie and Charles Vincent (Eds.). 2013. Proceedings of the 4th International Symposium on Biological Control of Arthropods. Pucón, Chile, 4-8 March 2013, 380 p. Proceedings of the 4th INTERNATIONAL SYMPOSIUM ON BIOLOGICAL CONTROL OF ARTHROPODS Pucón, Chile March 4-8, 2013 Peter G. Mason, David R. Gillespie and Charles Vincent (Eds.) 4th INTERNATIONAL SYMPOSIUM ON BIOLOGICAL CONTROL OF ARTHROPODS Pucón, Chile, March 4-8, 2013 PREFACE The Fourth International Symposium on Biological Control of Arthropods, held in Pucón – Chile, continues the series of international symposia on the biological control of arthropods organized every four years. The first meeting was in Hawaii – USA during January 2002, followed by the Davos - Switzerland meeting during September 2005, and the Christchurch – New Zealand meeting during February 2009. The goal of these symposia is to create a forum where biological control researchers and practitioners can meet and exchange information, to promote discussions of up to date issues affecting biological control, particularly pertaining to the use of parasitoids and predators as biological control agents.