Conformational Properties of Constrained Proline Analogues and Their Application in Nanobiology”
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Al Exposure Increases Proline Levels by Different Pathways in An
www.nature.com/scientificreports OPEN Al exposure increases proline levels by diferent pathways in an Al‑sensitive and an Al‑tolerant rye genotype Alexandra de Sousa1,2, Hamada AbdElgawad2,4, Fernanda Fidalgo1, Jorge Teixeira1, Manuela Matos3, Badreldin A. Hamed4, Samy Selim5, Wael N. Hozzein6, Gerrit T. S. Beemster2 & Han Asard2* Aluminium (Al) toxicity limits crop productivity, particularly at low soil pH. Proline (Pro) plays a role in protecting plants against various abiotic stresses. Using the relatively Al‑tolerant cereal rye (Secale cereale L.), we evaluated Pro metabolism in roots and shoots of two genotypes difering in Al tolerance, var. RioDeva (sensitive) and var. Beira (tolerant). Most enzyme activities and metabolites of Pro biosynthesis were analysed. Al induced increases in Pro levels in each genotype, but the mechanisms were diferent and were also diferent between roots and shoots. The Al‑tolerant genotype accumulated highest Pro levels and this stronger increase was ascribed to simultaneous activation of the ornithine (Orn)‑biosynthetic pathway and decrease in Pro oxidation. The Orn pathway was particularly enhanced in roots. Nitrate reductase (NR) activity, N levels, and N/C ratios demonstrate that N‑metabolism is less inhibited in the Al‑tolerant line. The correlation between Pro changes and diferences in Al‑sensitivity between these two genotypes, supports a role for Pro in Al tolerance. Our results suggest that diferential responses in Pro biosynthesis may be linked to N‑availability. Understanding the role of Pro in diferences between genotypes in stress responses, could be valuable in plant selection and breeding for Al resistance. Proline (Pro) is involved in a wide range of plant physiological and developmental processes1. -

The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn
biomolecules Review The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn 1, 1, 1 2 1,3 Daniel Rademaker y, Jarek van Dijk y, Willem Titulaer , Joanna Lange , Gert Vriend and Li Xue 1,* 1 Centre for Molecular and Biomolecular Informatics (CMBI), Radboudumc, 6525 GA Nijmegen, The Netherlands; [email protected] (D.R.); [email protected] (J.v.D.); [email protected] (W.T.); [email protected] (G.V.) 2 Bio-Prodict, 6511 AA Nijmegen, The Netherlands; [email protected] 3 Baco Institute of Protein Science (BIPS), Mindoro 5201, Philippines * Correspondence: [email protected] These authors contributed equally to this work. y Received: 15 May 2020; Accepted: 2 June 2020; Published: 16 June 2020 Abstract: When Oleg Ptitsyn and his group published the first secondary structure prediction for a protein sequence, they started a research field that is still active today. Oleg Ptitsyn combined fundamental rules of physics with human understanding of protein structures. Most followers in this field, however, use machine learning methods and aim at the highest (average) percentage correctly predicted residues in a set of proteins that were not used to train the prediction method. We show that one single method is unlikely to predict the secondary structure of all protein sequences, with the exception, perhaps, of future deep learning methods based on very large neural networks, and we suggest that some concepts pioneered by Oleg Ptitsyn and his group in the 70s of the previous century likely are today’s best way forward in the protein secondary structure prediction field. -

Amino Acid Recognition by Aminoacyl-Trna Synthetases
www.nature.com/scientificreports OPEN The structural basis of the genetic code: amino acid recognition by aminoacyl‑tRNA synthetases Florian Kaiser1,2,4*, Sarah Krautwurst3,4, Sebastian Salentin1, V. Joachim Haupt1,2, Christoph Leberecht3, Sebastian Bittrich3, Dirk Labudde3 & Michael Schroeder1 Storage and directed transfer of information is the key requirement for the development of life. Yet any information stored on our genes is useless without its correct interpretation. The genetic code defnes the rule set to decode this information. Aminoacyl-tRNA synthetases are at the heart of this process. We extensively characterize how these enzymes distinguish all natural amino acids based on the computational analysis of crystallographic structure data. The results of this meta-analysis show that the correct read-out of genetic information is a delicate interplay between the composition of the binding site, non-covalent interactions, error correction mechanisms, and steric efects. One of the most profound open questions in biology is how the genetic code was established. While proteins are encoded by nucleic acid blueprints, decoding this information in turn requires proteins. Te emergence of this self-referencing system poses a chicken-or-egg dilemma and its origin is still heavily debated 1,2. Aminoacyl-tRNA synthetases (aaRSs) implement the correct assignment of amino acids to their codons and are thus inherently connected to the emergence of genetic coding. Tese enzymes link tRNA molecules with their amino acid cargo and are consequently vital for protein biosynthesis. Beside the correct recognition of tRNA features3, highly specifc non-covalent interactions in the binding sites of aaRSs are required to correctly detect the designated amino acid4–7 and to prevent errors in biosynthesis5,8. -

Chapter 4 the Three-Dimensional Structure of Proteins
Chapter 4 The Three-Dimensional Structure of Proteins Multiple Choice Questions 1. Answer: D All of the following are considered “weak” interactions in proteins, except: A) hydrogen bonds. B) hydrophobic interactions. C) ionic bonds. D) peptide bonds. E) van der Waals forces. 2. Answer: D In an aqueous solution, protein conformation is determined by two major factors. One is the formation of the maximum number of hydrogen bonds. The other is the: A) formation of the maximum number of hydrophilic interactions. B) maximization of ionic interactions. C) minimization of entropy by the formation of a water solvent shell around the protein. D) placement of hydrophobic amino acid residues within the interior of the protein. E) placement of polar amino acid residues around the exterior of the protein. 3. 3 Answer: A In the diagram below, the plane drawn behind the peptide bond indicates the: A) absence of rotation around the C—N bond because of its partial double-bond character. B) plane of rotation around the C—N bond. C) region of steric hindrance determined by the large C=O group. D) region of the peptide bond that contributes to a Ramachandran plot. E) theoretical space between –180 and +180 degrees that can be occupied by the and angles in the peptide bond. 4. Answer: D Which of the following best represents the backbone arrangement of two peptide bonds? A) C—N—C—C—C—N—C—C B) C—N—C—C—N—C C) C—N—C—C—C—N D) C—C—N—C—C—N Chapter 4 The Three-Dimensional Structure of Proteins E) C—C—C—N—C—C—C 5. -

2. Proteins Have Hierarchies of Structure
27 2. Proteins have Hierarchies of Structure Protein structure is usually described at four different levels (Fig. II.2.1). The first level, called the primary structure, describes the linear sequence of the amino acids in the chain. The different primary structures correspond to the different sequences in which the amino acids are covalently linked together. The secondary structure describes two common patterns of structural repetition in proteins: the coiling up into helices of segments of the chain, and the pairing together of strands of the chain into β-sheets. The tertiary structure is the next higher level of organization, the overall arrangement of secondary structural elements. The quaternary structure describes how different polypeptide chains are assembled into complexes. Figure II.2.1. Different levels of protein structure. A protein chain’s primary structure is its amino acid sequence. Secondary structure consists of the regular organization of helices and sheets. The example shown is a schematic representation of an α helix. Tertiary structure is a polypeptide chain’s three- dimensional native conformation, which often involves compact packing of secondary structure elements. The example given in the figure is a schematic drawing of one of the four polypeptide chains (subunits) of hemoglobin, the protein that transports oxygen in the blood. α-helices along the chain are represented as cylinders. N is the amino terminus and C is the carboxyl terminus of the polypeptide chain. Quaternary structure is the arrangement of multiple polypeptide chains (subunits) to form a functional biomolecular structure. The figure shows the quaternary arrangement of four subunits to form the functional hemoglobin molecule. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Detection of Trans-Cis Flips and Peptide-Plane Flips in Protein Structures
research papers Detection of trans–cis flips and peptide-plane flips in protein structures ISSN 1399-0047 Wouter G. Touw,a* Robbie P. Joostenb and Gert Vrienda* aCentre for Molecular and Biomolecular Informatics, Radboud University Medical Center, Geert Grooteplein-Zuid 26-28, 6525 GA Nijmegen, The Netherlands, and bDepartment of Biochemistry, Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. *Correspondence e-mail: [email protected], Received 4 February 2015 [email protected] Accepted 27 April 2015 A coordinate-based method is presented to detect peptide bonds that need Edited by G. J. Kleywegt, EMBL–EBI, Hinxton, correction either by a peptide-plane flip or by a trans–cis inversion of the peptide England bond. When applied to the whole Protein Data Bank, the method predicts 4617 trans–cis flips and many thousands of hitherto unknown peptide-plane flips. Keywords: peptide conformation; cis peptide A few examples are highlighted for which a correction of the peptide-plane bond; structure validation; structure correction. geometry leads to a correction of the understanding of the structure–function relation. All data, including 1088 manually validated cases, are freely available and the method is available from a web server, a web-service interface and through WHAT_CHECK. 1. Introduction Peptide bonds connect adjacent amino acids in proteins. The partial double-bond character of the peptide bond restricts its torsion. The dihedral angle ! (CiÀ1—CiÀ1—Ni—Ci ) typically has values around 180 (trans)or0 (cis), although exceptions are possible (Berkholz et al., 2012). The Ci–1—Ci distance is around 3.81 A˚ in the trans conformation and around 2.94 A˚ in the cis conformation. -
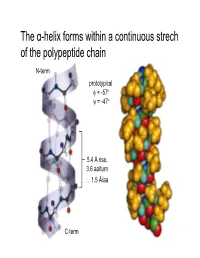
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Dietary and Pharmacological Induction of Serine Synthesis Genes
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.15.151860; this version posted June 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Dietary and pharmacological induction of serine synthesis genes Alexei Vazquez1,2,* 1Cancer Research UK Beatson Institute, Glasgow, UK 2Institute of Cancer Sciences, University of Glasgow, Glasgow, UK Email: [email protected] Abstract gene coding for 3-phosphoglycerate dehydrogenase (Phgdh), the first enzyme of serine synthesis, causes There is an increasing interest in the pathway of L-serine neurodevelopmental abnormalities and it is embryonic synthesis and its. Although L-serine and downstream lethal 4 in mice. products can be obtained from the diet, serine deficiency has been documented in neurological disorders, macular The manifestation of serine deficiency in the human degeneration and aging. This evidence calls for strategies population call for strategies to increase the availability of to induce serine synthesis. Here I address this problem serine. Dietary serine supplementation seems the obvious taking advantage of the wealth of data deposited in the strategy 5. However, for a number of reasons, serine gene expression omnibus database. I uncover that low supplementation is not an effective approach in general 5. protein and ketogenic diets increase the expression of Dietary serine is rapidly incorporated into proteins or serine synthesis genes in the liver and the brain relative to broken down to glycine and formate 6. -

And Beta-Helical Protein Motifs
Soft Matter Mechanical Unfolding of Alpha- and Beta-helical Protein Motifs Journal: Soft Matter Manuscript ID SM-ART-10-2018-002046.R1 Article Type: Paper Date Submitted by the 28-Nov-2018 Author: Complete List of Authors: DeBenedictis, Elizabeth; Northwestern University Keten, Sinan; Northwestern University, Mechanical Engineering Page 1 of 10 Please doSoft not Matter adjust margins Soft Matter ARTICLE Mechanical Unfolding of Alpha- and Beta-helical Protein Motifs E. P. DeBenedictis and S. Keten* Received 24th September 2018, Alpha helices and beta sheets are the two most common secondary structure motifs in proteins. Beta-helical structures Accepted 00th January 20xx merge features of the two motifs, containing two or three beta-sheet faces connected by loops or turns in a single protein. Beta-helical structures form the basis of proteins with diverse mechanical functions such as bacterial adhesins, phage cell- DOI: 10.1039/x0xx00000x puncture devices, antifreeze proteins, and extracellular matrices. Alpha helices are commonly found in cellular and extracellular matrix components, whereas beta-helices such as curli fibrils are more common as bacterial and biofilm matrix www.rsc.org/ components. It is currently not known whether it may be advantageous to use one helical motif over the other for different structural and mechanical functions. To better understand the mechanical implications of using different helix motifs in networks, here we use Steered Molecular Dynamics (SMD) simulations to mechanically unfold multiple alpha- and beta- helical proteins at constant velocity at the single molecule scale. We focus on the energy dissipated during unfolding as a means of comparison between proteins and work normalized by protein characteristics (initial and final length, # H-bonds, # residues, etc.). -

PA28: New Insights on an Ancient Proteasome Activator
biomolecules Review PA28γ: New Insights on an Ancient Proteasome Activator Paolo Cascio Department of Veterinary Sciences, University of Turin, Largo P. Braccini 2, 10095 Grugliasco, Italy; [email protected] Abstract: PA28 (also known as 11S, REG or PSME) is a family of proteasome regulators whose members are widely present in many of the eukaryotic supergroups. In jawed vertebrates they are represented by three paralogs, PA28α, PA28β, and PA28γ, which assemble as heptameric hetero (PA28αβ) or homo (PA28γ) rings on one or both extremities of the 20S proteasome cylindrical structure. While they share high sequence and structural similarities, the three isoforms significantly differ in terms of their biochemical and biological properties. In fact, PA28α and PA28β seem to have appeared more recently and to have evolved very rapidly to perform new functions that are specifically aimed at optimizing the process of MHC class I antigen presentation. In line with this, PA28αβ favors release of peptide products by proteasomes and is particularly suited to support adaptive immune responses without, however, affecting hydrolysis rates of protein substrates. On the contrary, PA28γ seems to be a slow-evolving gene that is most similar to the common ancestor of the PA28 activators family, and very likely retains its original functions. Notably, PA28γ has a prevalent nuclear localization and is involved in the regulation of several essential cellular processes including cell growth and proliferation, apoptosis, chromatin structure and organization, and response to DNA damage. In striking contrast with the activity of PA28αβ, most of these diverse biological functions of PA28γ seem to depend on its ability to markedly enhance degradation rates of regulatory protein by 20S proteasome. -

Nonproteinogenic Deep Mutational Scanning of Linear and Cyclic Peptides
Nonproteinogenic deep mutational scanning of linear and cyclic peptides Joseph M. Rogersa, Toby Passiouraa, and Hiroaki Sugaa,b,1 aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, Tokyo 113-0033, Japan; and bCore Research for Evolutionary Science and Technology, Japan Science and Technology Agency, Saitama 332-0012, Japan Edited by David Baker, University of Washington, Seattle, WA, and approved September 18, 2018 (received for review June 10, 2018) High-resolution structure–activity analysis of polypeptides re- mutants that can be constructed (18). Moreover, it is possible to quires amino acid structures that are not present in the universal combine parallel peptide synthesis with measures of function genetic code. Examination of peptide and protein interactions (19). However, these approaches cannot construct peptide li- with this resolution has been limited by the need to individually braries with the sequence length and numbers that deep muta- synthesize and test peptides containing nonproteinogenic amino tional scanning can, which, at its core, uses high-fidelity nucleic acids. We describe a method to scan entire peptide sequences with acid-directed synthesis of polypeptides by the ribosome. multiple nonproteinogenic amino acids and, in parallel, determine Ribosomal synthesis (i.e., translation) can be manipulated to the thermodynamics of binding to a partner protein. By coupling include nonproteinogenic amino acids (20). In vitro genetic code genetic code reprogramming to deep mutational scanning, any reprogramming is particularly versatile, allowing for the in- number of amino acids can be exhaustively substituted into pep- corporation of amino acids with diverse chemical structures (21). tides, and single experiments can return all free energy changes of Flexizymes, flexible tRNA-acylation ribozymes, can load almost binding.