2. Proteins Have Hierarchies of Structure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Side-Chain Liquid Crystal Polymers (SCLCP): Methods and Materials. an Overview
Materials 2009, 2, 95-128; doi:10.3390/ma2010095 OPEN ACCESS materials ISSN 1996-1944 www.mdpi.com/journal/materials Review Side-chain Liquid Crystal Polymers (SCLCP): Methods and Materials. An Overview Tomasz Ganicz * and Włodzimierz Stańczyk Centre of Molecular and Macromolecular Studies, Polish Academy of Science, Sienkiewicza 112, 90- 363 Łódź, Poland; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel. +48-42-684-71-13; Fax: +48-42-684-71-26 Received: 5 February 2009; in revised form: 3 March 2009 / Accepted: 9 March 2009 / Published: 11 March 2009 Abstract: This review focuses on recent developments in the chemistry of side chain liquid crystal polymers. It concentrates on current trends in synthetic methods and novel, well defined structures, supramolecular arrangements, properties, and applications. The review covers literature published in this century, apart from some areas, such as dendritic and elastomeric systems, which have been recently reviewed. Keywords: Liquid crystals, side chain polymers, polymer modification, polymer synthesis, self-assembly. 1. Introduction Over thirty years ago the work of Finkelmann and Ringsdorf [1,2] gave important momentum to synthesis of side chain liquid crystal polymers (SCLCPs), materials which combine the anisotropy of liquid crystalline mesogens with the mechanical properties of polymers. Although there were some earlier attempts [2], their approach of decoupling the motions of a polymer main chain from a mesogen, thus allowed side chain moieties to build up long range ordering (Figure 1). They were able to synthesize polymers with nematic, smectic and cholesteric phases via free radical polymerization of methacryloyl type monomers [3]. -

The Role of Side-Chain Branch Position on Thermal Property of Poly-3-Alkylthiophenes
Polymer Chemistry The Role of Side-chain Branch Position on Thermal Property of Poly-3-alkylthiophenes Journal: Polymer Chemistry Manuscript ID PY-ART-07-2019-001026.R2 Article Type: Paper Date Submitted by the 02-Oct-2019 Author: Complete List of Authors: Gu, Xiaodan; University of Southern Mississippi, School of Polymer Science and Engineering Cao, Zhiqiang; University of Southern Mississippi, School of Polymer Science and Engineering Galuska, Luke; University of Southern Mississippi, School of Polymer Science and Engineering Qian, Zhiyuan; University of Southern Mississippi, School of Polymer Science and Engineering Zhang, Song; University of Southern Mississippi, School of Polymer Science and Engineering Huang, Lifeng; University of Southern Mississippi, School of Polymers and High Performance Materials Prine, Nathaniel; University of Southern Mississippi, School of Polymer Science and Engineering Li, Tianyu; Oak Ridge National Laboratory; University of Tennessee Knoxville He, Youjun; Oak Ridge National Laboratory Hong, Kunlun; Oak Ridge National Laboratory, Center for Nanophase Materials Science; Oak Ridge National Laboratory, Center for Nanophase Materials Science Page 1 of 13 PleasePolymer do not Chemistryadjust margins ARTICLE The Role of Side-chain Branch Position on Thermal Property of Poly-3-alkylthiophenes Received 00th January 20xx, a a a a a a Accepted 00th January 20xx Zhiqiang Cao, Luke Galuska, Zhiyuan Qian, Song Zhang, Lifeng Huang, Nathaniel Prine, Tianyu Li,b, c Youjun He,b Kunlun Hong,*b, c and Xiaodan Gu*a DOI: 10.1039/x0xx00000x Thermomechanical properties of conjugated polymers (CPs) are greatly influenced by both their microstructures and backbone structures. In the present work, to investigate the effect of side-chain branch position on the backbone’s mobility and molecular packing structure, four poly (3-alkylthiophene-2, 5-diyl) derivatives (P3ATs) with different side chains, either branched and linear, were synthesized by a quasi-living Kumada catalyst transfer polymerization (KCTP) method. -

Improved Prediction of Protein Side-Chain Conformations with SCWRL4 Georgii G
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Improved prediction of protein side-chain conformations with SCWRL4 Georgii G. Krivov,1,2 Maxim V. Shapovalov,1 and Roland L. Dunbrack Jr.1* 1 Institute for Cancer Research, Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia, Pennsylvania 19111 2 Department of Applied Mathematics, Moscow Engineering Physics Institute (MEPhI), Kashirskoe Shosse 31, Moscow 115409, Russian Federation INTRODUCTION ABSTRACT The side-chain conformation prediction problem is an integral Determination of side-chain conformations is an component of protein structure determination, protein structure important step in protein structure prediction and protein design. Many such methods have prediction, and protein design. In single-site mutants and in closely been presented, although only a small number related proteins, the backbone often changes little and structure pre- 1 are in widespread use. SCWRL is one such diction can be accomplished by accurate side-chain prediction. In method, and the SCWRL3 program (2003) has docking of ligands and other proteins, taking into account changes remained popular because of its speed, accuracy, in side-chain conformation is often critical to accurate structure pre- and ease-of-use for the purpose of homology dictions of complexes.2–4 Even in methods that take account of modeling. However, higher accuracy at compara- changes in backbone conformation, one step in the process is recal- ble speed is desirable. This has been achieved in culation of side-chain conformation or ‘‘repacking.’’5 Because many a new program SCWRL4 through: (1) a new backbone conformations may be sampled in model refinements, backbone-dependent rotamer library based on side-chain prediction must also be very fast. -

The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn
biomolecules Review The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn 1, 1, 1 2 1,3 Daniel Rademaker y, Jarek van Dijk y, Willem Titulaer , Joanna Lange , Gert Vriend and Li Xue 1,* 1 Centre for Molecular and Biomolecular Informatics (CMBI), Radboudumc, 6525 GA Nijmegen, The Netherlands; [email protected] (D.R.); [email protected] (J.v.D.); [email protected] (W.T.); [email protected] (G.V.) 2 Bio-Prodict, 6511 AA Nijmegen, The Netherlands; [email protected] 3 Baco Institute of Protein Science (BIPS), Mindoro 5201, Philippines * Correspondence: [email protected] These authors contributed equally to this work. y Received: 15 May 2020; Accepted: 2 June 2020; Published: 16 June 2020 Abstract: When Oleg Ptitsyn and his group published the first secondary structure prediction for a protein sequence, they started a research field that is still active today. Oleg Ptitsyn combined fundamental rules of physics with human understanding of protein structures. Most followers in this field, however, use machine learning methods and aim at the highest (average) percentage correctly predicted residues in a set of proteins that were not used to train the prediction method. We show that one single method is unlikely to predict the secondary structure of all protein sequences, with the exception, perhaps, of future deep learning methods based on very large neural networks, and we suggest that some concepts pioneered by Oleg Ptitsyn and his group in the 70s of the previous century likely are today’s best way forward in the protein secondary structure prediction field. -

Molecular Modelling Virtual Chemistry Resource Pymol Experiment Answer Sheet Well Done for Completing Our Pymol Experiment! We Really Hope You Enjoyed It!
#Outtheboxthinking Faculty of Natural & Mathematical Sciences Department of Chemistry Molecular Modelling Virtual Chemistry Resource PyMol Experiment Answer Sheet Well done for completing our PyMol experiment! We really hope you enjoyed it! Please make sure you have completed our post experiment questionnaire. https://kings.onlinesurveys.ac.uk/post-questionnaire-for-outtheboxthinking-resources Send us photos, comments, or thoughts to either our twitter or Instagram accounts using the hashtag #outtheboxthinking. We also encourage you to make a poster of your data and email it to us. Exploring how proteins are formed Press “Amino Acid” Q. What is the R group in the side chain of this amino acid? CH3- Methyl Group Q. Which amino acid is shown here? Alanine Press the button “ALA_ASP” and an additional amino acid will appear Q. What is the chemical formula of the side chain of the new amino acid? CH2COOH Q. What is the name of the second, new amino acid? Aspartic Acid or Aspartate Press the button “Bonded_Ala_Asp” Q. What colour(s) is the amide bond connecting the two amino acids? Blue and green. It is important to highlight the bond between the nitrogen of one amino acid and the carbonyl of the second amino acid and not any other bond (Figure 1). Figure 1: Arrow points to the amide bond formed between two amino acids. Page 1 of 8 Instagram: kclchemoutreach #outtheboxthinking Q. Which elements have been lost from each amino acid following their bonding? Amino acid 1:: Alanine has lost an oxygen and a hydrogen Amino acid 2: Aspartic Acid has lost a hydrogen If you are unsure why this is correct look at figure 2 and open the PyMol document again to visualise the bonding Figure 2: Reaction scheme for amino acid bonding, resulting in the formation of an amide bond and the loss of water. -

Amino Acid Recognition by Aminoacyl-Trna Synthetases
www.nature.com/scientificreports OPEN The structural basis of the genetic code: amino acid recognition by aminoacyl‑tRNA synthetases Florian Kaiser1,2,4*, Sarah Krautwurst3,4, Sebastian Salentin1, V. Joachim Haupt1,2, Christoph Leberecht3, Sebastian Bittrich3, Dirk Labudde3 & Michael Schroeder1 Storage and directed transfer of information is the key requirement for the development of life. Yet any information stored on our genes is useless without its correct interpretation. The genetic code defnes the rule set to decode this information. Aminoacyl-tRNA synthetases are at the heart of this process. We extensively characterize how these enzymes distinguish all natural amino acids based on the computational analysis of crystallographic structure data. The results of this meta-analysis show that the correct read-out of genetic information is a delicate interplay between the composition of the binding site, non-covalent interactions, error correction mechanisms, and steric efects. One of the most profound open questions in biology is how the genetic code was established. While proteins are encoded by nucleic acid blueprints, decoding this information in turn requires proteins. Te emergence of this self-referencing system poses a chicken-or-egg dilemma and its origin is still heavily debated 1,2. Aminoacyl-tRNA synthetases (aaRSs) implement the correct assignment of amino acids to their codons and are thus inherently connected to the emergence of genetic coding. Tese enzymes link tRNA molecules with their amino acid cargo and are consequently vital for protein biosynthesis. Beside the correct recognition of tRNA features3, highly specifc non-covalent interactions in the binding sites of aaRSs are required to correctly detect the designated amino acid4–7 and to prevent errors in biosynthesis5,8. -

Chapter 4 the Three-Dimensional Structure of Proteins
Chapter 4 The Three-Dimensional Structure of Proteins Multiple Choice Questions 1. Answer: D All of the following are considered “weak” interactions in proteins, except: A) hydrogen bonds. B) hydrophobic interactions. C) ionic bonds. D) peptide bonds. E) van der Waals forces. 2. Answer: D In an aqueous solution, protein conformation is determined by two major factors. One is the formation of the maximum number of hydrogen bonds. The other is the: A) formation of the maximum number of hydrophilic interactions. B) maximization of ionic interactions. C) minimization of entropy by the formation of a water solvent shell around the protein. D) placement of hydrophobic amino acid residues within the interior of the protein. E) placement of polar amino acid residues around the exterior of the protein. 3. 3 Answer: A In the diagram below, the plane drawn behind the peptide bond indicates the: A) absence of rotation around the C—N bond because of its partial double-bond character. B) plane of rotation around the C—N bond. C) region of steric hindrance determined by the large C=O group. D) region of the peptide bond that contributes to a Ramachandran plot. E) theoretical space between –180 and +180 degrees that can be occupied by the and angles in the peptide bond. 4. Answer: D Which of the following best represents the backbone arrangement of two peptide bonds? A) C—N—C—C—C—N—C—C B) C—N—C—C—N—C C) C—N—C—C—C—N D) C—C—N—C—C—N Chapter 4 The Three-Dimensional Structure of Proteins E) C—C—C—N—C—C—C 5. -

Conformational Properties of Constrained Proline Analogues and Their Application in Nanobiology”
UNIVERSITAT POLITÈCNICA DE CATALUNYA DEPARTAMENT D’ENGINYERIA QUÍMICA “CONFORMATIONAL PROPERTIES OF CONSTRAINED PROLINE ANALOGUES AND THEIR APPLICATION IN NANOBIOLOGY” Alejandra Flores Ortega Supervisors: Dr. Carlos Alemán Llansó and Dr. Jordi Casanovas Salas. th Barcelona, 27 January 2009 “Chance is a word void of sense; nothing can exist without a cause”. François-Marie Arouet, Voltaire “Imagination will often carry us to worlds that never were. But without it, we go nowhere”. Carl Sagan iii ACKNOWLEDGEMENTS I would like to acknowledge to Dr. Carlos Aleman and Dr. Jordi Cassanovas Salas for an interesting research theme, and scientific support. I gratefully acknowledge to Dr. David Zanuy for interesting suggestions and strong discussions, without their support this would be an unfulfilled task. Also I, would like to address my thanks to all my colleagues in my group and department, specially Elaine Armelin for assiting me in many different ways. I thank not only my friends, but also colleagues for helping me to overcome the stressful time, without whom it would have been difficult to cope up. I wish to express my gratefulness to my parents, specially to my mother, María Esther, for all his care, and support. Also I will like to thanks to my friends and specially Jesus, Merches, Laura y Arturo. My PhD thesis have been finished for all this support. I am greatly indepted to Dr. Ruth Nussinov at NCI, Dr. Carlos Cativiela at the University of Zaragoza and Ana I. Jiménez at the “Instituto de Ciencias de Materiales de Aragon” for a collaborative effort. I wish to thank all my colleague in the “Chimie et Biochimie Théoriques, Faculté des Sciences et Techniques” in Nancy France, I will be grateful to have worked with : Pr. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Detection of Trans-Cis Flips and Peptide-Plane Flips in Protein Structures
research papers Detection of trans–cis flips and peptide-plane flips in protein structures ISSN 1399-0047 Wouter G. Touw,a* Robbie P. Joostenb and Gert Vrienda* aCentre for Molecular and Biomolecular Informatics, Radboud University Medical Center, Geert Grooteplein-Zuid 26-28, 6525 GA Nijmegen, The Netherlands, and bDepartment of Biochemistry, Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. *Correspondence e-mail: [email protected], Received 4 February 2015 [email protected] Accepted 27 April 2015 A coordinate-based method is presented to detect peptide bonds that need Edited by G. J. Kleywegt, EMBL–EBI, Hinxton, correction either by a peptide-plane flip or by a trans–cis inversion of the peptide England bond. When applied to the whole Protein Data Bank, the method predicts 4617 trans–cis flips and many thousands of hitherto unknown peptide-plane flips. Keywords: peptide conformation; cis peptide A few examples are highlighted for which a correction of the peptide-plane bond; structure validation; structure correction. geometry leads to a correction of the understanding of the structure–function relation. All data, including 1088 manually validated cases, are freely available and the method is available from a web server, a web-service interface and through WHAT_CHECK. 1. Introduction Peptide bonds connect adjacent amino acids in proteins. The partial double-bond character of the peptide bond restricts its torsion. The dihedral angle ! (CiÀ1—CiÀ1—Ni—Ci ) typically has values around 180 (trans)or0 (cis), although exceptions are possible (Berkholz et al., 2012). The Ci–1—Ci distance is around 3.81 A˚ in the trans conformation and around 2.94 A˚ in the cis conformation. -
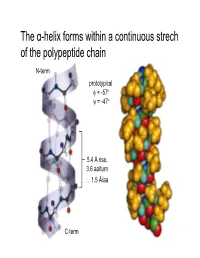
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

A Switch Between Two-, Three-, and Four-Stranded Coiled Coils in GCN4 Leucine Zipper Mutants
A Switch Between Two-, Three-, and Four-Stranded Coiled Coils in GCN4 Leucine Zipper Mutants Pehr B. Harbury; Tao Zhang; Peter S. Kim; Tom Alber Science, New Series, Vol. 262, No. 5138. (Nov. 26, 1993), pp. 1401-1407. Stable URL: http://links.jstor.org/sici?sici=0036-8075%2819931126%293%3A262%3A5138%3C1401%3AASBTTA%3E2.0.CO%3B2-H Science is currently published by American Association for the Advancement of Science. Your use of the JSTOR archive indicates your acceptance of JSTOR's Terms and Conditions of Use, available at http://www.jstor.org/about/terms.html. JSTOR's Terms and Conditions of Use provides, in part, that unless you have obtained prior permission, you may not download an entire issue of a journal or multiple copies of articles, and you may use content in the JSTOR archive only for your personal, non-commercial use. Please contact the publisher regarding any further use of this work. Publisher contact information may be obtained at http://www.jstor.org/journals/aaas.html. Each copy of any part of a JSTOR transmission must contain the same copyright notice that appears on the screen or printed page of such transmission. The JSTOR Archive is a trusted digital repository providing for long-term preservation and access to leading academic journals and scholarly literature from around the world. The Archive is supported by libraries, scholarly societies, publishers, and foundations. It is an initiative of JSTOR, a not-for-profit organization with a mission to help the scholarly community take advantage of advances in technology. For more information regarding JSTOR, please contact [email protected].