Detection of Trans-Cis Flips and Peptide-Plane Flips in Protein Structures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn
biomolecules Review The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn 1, 1, 1 2 1,3 Daniel Rademaker y, Jarek van Dijk y, Willem Titulaer , Joanna Lange , Gert Vriend and Li Xue 1,* 1 Centre for Molecular and Biomolecular Informatics (CMBI), Radboudumc, 6525 GA Nijmegen, The Netherlands; [email protected] (D.R.); [email protected] (J.v.D.); [email protected] (W.T.); [email protected] (G.V.) 2 Bio-Prodict, 6511 AA Nijmegen, The Netherlands; [email protected] 3 Baco Institute of Protein Science (BIPS), Mindoro 5201, Philippines * Correspondence: [email protected] These authors contributed equally to this work. y Received: 15 May 2020; Accepted: 2 June 2020; Published: 16 June 2020 Abstract: When Oleg Ptitsyn and his group published the first secondary structure prediction for a protein sequence, they started a research field that is still active today. Oleg Ptitsyn combined fundamental rules of physics with human understanding of protein structures. Most followers in this field, however, use machine learning methods and aim at the highest (average) percentage correctly predicted residues in a set of proteins that were not used to train the prediction method. We show that one single method is unlikely to predict the secondary structure of all protein sequences, with the exception, perhaps, of future deep learning methods based on very large neural networks, and we suggest that some concepts pioneered by Oleg Ptitsyn and his group in the 70s of the previous century likely are today’s best way forward in the protein secondary structure prediction field. -

Chapter 4 the Three-Dimensional Structure of Proteins
Chapter 4 The Three-Dimensional Structure of Proteins Multiple Choice Questions 1. Answer: D All of the following are considered “weak” interactions in proteins, except: A) hydrogen bonds. B) hydrophobic interactions. C) ionic bonds. D) peptide bonds. E) van der Waals forces. 2. Answer: D In an aqueous solution, protein conformation is determined by two major factors. One is the formation of the maximum number of hydrogen bonds. The other is the: A) formation of the maximum number of hydrophilic interactions. B) maximization of ionic interactions. C) minimization of entropy by the formation of a water solvent shell around the protein. D) placement of hydrophobic amino acid residues within the interior of the protein. E) placement of polar amino acid residues around the exterior of the protein. 3. 3 Answer: A In the diagram below, the plane drawn behind the peptide bond indicates the: A) absence of rotation around the C—N bond because of its partial double-bond character. B) plane of rotation around the C—N bond. C) region of steric hindrance determined by the large C=O group. D) region of the peptide bond that contributes to a Ramachandran plot. E) theoretical space between –180 and +180 degrees that can be occupied by the and angles in the peptide bond. 4. Answer: D Which of the following best represents the backbone arrangement of two peptide bonds? A) C—N—C—C—C—N—C—C B) C—N—C—C—N—C C) C—N—C—C—C—N D) C—C—N—C—C—N Chapter 4 The Three-Dimensional Structure of Proteins E) C—C—C—N—C—C—C 5. -

Conformational Properties of Constrained Proline Analogues and Their Application in Nanobiology”
UNIVERSITAT POLITÈCNICA DE CATALUNYA DEPARTAMENT D’ENGINYERIA QUÍMICA “CONFORMATIONAL PROPERTIES OF CONSTRAINED PROLINE ANALOGUES AND THEIR APPLICATION IN NANOBIOLOGY” Alejandra Flores Ortega Supervisors: Dr. Carlos Alemán Llansó and Dr. Jordi Casanovas Salas. th Barcelona, 27 January 2009 “Chance is a word void of sense; nothing can exist without a cause”. François-Marie Arouet, Voltaire “Imagination will often carry us to worlds that never were. But without it, we go nowhere”. Carl Sagan iii ACKNOWLEDGEMENTS I would like to acknowledge to Dr. Carlos Aleman and Dr. Jordi Cassanovas Salas for an interesting research theme, and scientific support. I gratefully acknowledge to Dr. David Zanuy for interesting suggestions and strong discussions, without their support this would be an unfulfilled task. Also I, would like to address my thanks to all my colleagues in my group and department, specially Elaine Armelin for assiting me in many different ways. I thank not only my friends, but also colleagues for helping me to overcome the stressful time, without whom it would have been difficult to cope up. I wish to express my gratefulness to my parents, specially to my mother, María Esther, for all his care, and support. Also I will like to thanks to my friends and specially Jesus, Merches, Laura y Arturo. My PhD thesis have been finished for all this support. I am greatly indepted to Dr. Ruth Nussinov at NCI, Dr. Carlos Cativiela at the University of Zaragoza and Ana I. Jiménez at the “Instituto de Ciencias de Materiales de Aragon” for a collaborative effort. I wish to thank all my colleague in the “Chimie et Biochimie Théoriques, Faculté des Sciences et Techniques” in Nancy France, I will be grateful to have worked with : Pr. -

2. Proteins Have Hierarchies of Structure
27 2. Proteins have Hierarchies of Structure Protein structure is usually described at four different levels (Fig. II.2.1). The first level, called the primary structure, describes the linear sequence of the amino acids in the chain. The different primary structures correspond to the different sequences in which the amino acids are covalently linked together. The secondary structure describes two common patterns of structural repetition in proteins: the coiling up into helices of segments of the chain, and the pairing together of strands of the chain into β-sheets. The tertiary structure is the next higher level of organization, the overall arrangement of secondary structural elements. The quaternary structure describes how different polypeptide chains are assembled into complexes. Figure II.2.1. Different levels of protein structure. A protein chain’s primary structure is its amino acid sequence. Secondary structure consists of the regular organization of helices and sheets. The example shown is a schematic representation of an α helix. Tertiary structure is a polypeptide chain’s three- dimensional native conformation, which often involves compact packing of secondary structure elements. The example given in the figure is a schematic drawing of one of the four polypeptide chains (subunits) of hemoglobin, the protein that transports oxygen in the blood. α-helices along the chain are represented as cylinders. N is the amino terminus and C is the carboxyl terminus of the polypeptide chain. Quaternary structure is the arrangement of multiple polypeptide chains (subunits) to form a functional biomolecular structure. The figure shows the quaternary arrangement of four subunits to form the functional hemoglobin molecule. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -
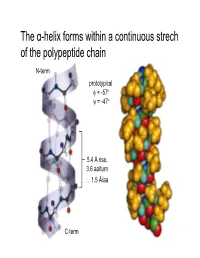
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

And Beta-Helical Protein Motifs
Soft Matter Mechanical Unfolding of Alpha- and Beta-helical Protein Motifs Journal: Soft Matter Manuscript ID SM-ART-10-2018-002046.R1 Article Type: Paper Date Submitted by the 28-Nov-2018 Author: Complete List of Authors: DeBenedictis, Elizabeth; Northwestern University Keten, Sinan; Northwestern University, Mechanical Engineering Page 1 of 10 Please doSoft not Matter adjust margins Soft Matter ARTICLE Mechanical Unfolding of Alpha- and Beta-helical Protein Motifs E. P. DeBenedictis and S. Keten* Received 24th September 2018, Alpha helices and beta sheets are the two most common secondary structure motifs in proteins. Beta-helical structures Accepted 00th January 20xx merge features of the two motifs, containing two or three beta-sheet faces connected by loops or turns in a single protein. Beta-helical structures form the basis of proteins with diverse mechanical functions such as bacterial adhesins, phage cell- DOI: 10.1039/x0xx00000x puncture devices, antifreeze proteins, and extracellular matrices. Alpha helices are commonly found in cellular and extracellular matrix components, whereas beta-helices such as curli fibrils are more common as bacterial and biofilm matrix www.rsc.org/ components. It is currently not known whether it may be advantageous to use one helical motif over the other for different structural and mechanical functions. To better understand the mechanical implications of using different helix motifs in networks, here we use Steered Molecular Dynamics (SMD) simulations to mechanically unfold multiple alpha- and beta- helical proteins at constant velocity at the single molecule scale. We focus on the energy dissipated during unfolding as a means of comparison between proteins and work normalized by protein characteristics (initial and final length, # H-bonds, # residues, etc.). -

PA28: New Insights on an Ancient Proteasome Activator
biomolecules Review PA28γ: New Insights on an Ancient Proteasome Activator Paolo Cascio Department of Veterinary Sciences, University of Turin, Largo P. Braccini 2, 10095 Grugliasco, Italy; [email protected] Abstract: PA28 (also known as 11S, REG or PSME) is a family of proteasome regulators whose members are widely present in many of the eukaryotic supergroups. In jawed vertebrates they are represented by three paralogs, PA28α, PA28β, and PA28γ, which assemble as heptameric hetero (PA28αβ) or homo (PA28γ) rings on one or both extremities of the 20S proteasome cylindrical structure. While they share high sequence and structural similarities, the three isoforms significantly differ in terms of their biochemical and biological properties. In fact, PA28α and PA28β seem to have appeared more recently and to have evolved very rapidly to perform new functions that are specifically aimed at optimizing the process of MHC class I antigen presentation. In line with this, PA28αβ favors release of peptide products by proteasomes and is particularly suited to support adaptive immune responses without, however, affecting hydrolysis rates of protein substrates. On the contrary, PA28γ seems to be a slow-evolving gene that is most similar to the common ancestor of the PA28 activators family, and very likely retains its original functions. Notably, PA28γ has a prevalent nuclear localization and is involved in the regulation of several essential cellular processes including cell growth and proliferation, apoptosis, chromatin structure and organization, and response to DNA damage. In striking contrast with the activity of PA28αβ, most of these diverse biological functions of PA28γ seem to depend on its ability to markedly enhance degradation rates of regulatory protein by 20S proteasome. -

Bending, Twisting and Turning: Protein Modeling and Visualization
List of papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I U. H. Danielsson, M. Lundgren and A. J. Niemi, Gauge field theory of chirally folded homopolymers with applications to folded proteins, Phys. Rev. E 82, 021910 (2010) II M. N. Chernodub, M. Lundgren and A. J. Niemi, Elastic energy and phase structure in a continuous spin Ising chain with applications to chiral homopolymers, Phys. Rev. E 83, 011126 (2011) III S. Hu, M. Lundgren and A. J. Niemi, Discrete Frenet frame, inflection point solitons, and curve visualization with applications to folded proteins, Phys. Rev. E 83 061908 (2011) IV M. Lundgren, A. J. Niemi and F. Sha, Protein loops, solitons and side-chain visualization with applications to the left-handed helix region, arXiv:1104.2261 [q-bio.BM]. Submitted to Phys. Rev. E. V M. Lundgren and A. J. Niemi, Backbone covalent bond dynamical symmetry breaking and side-chain geometry of folded proteins, arXiv:1109.0423 [q-bio.BM]. Submitted to Phys. Rev. E. Reprints were made with permission from the publishers. Contents 1 Introduction and background ...................................................................... 7 1.1 A quick guide to protein folding ..................................................... 7 1.2 Outline ............................................................................................ 15 Part I: Protein Geometry and Visualization 2 Main chain geometry ................................................................................. 19 2.1 -

Predicting Protein Secondary and Supersecondary Structure
29 Predicting Protein Secondary and Supersecondary Structure 29.1 Introduction............................................ 29-1 Background • Difficulty of general protein structure prediction • A bottom-up approach 29.2 Secondary structure ................................... 29-5 Early approaches • Incorporating local dependencies • Exploiting evolutionary information • Recent developments and conclusions 29.3 Tight turns ............................................. 29-13 29.4 Beta hairpins........................................... 29-15 29.5 Coiled coils ............................................. 29-16 Early approaches • Incorporating local dependencies • Predicting oligomerization • Structure-based predictions • Predicting coiled-coil protein interactions Mona Singh • Promising future directions Princeton University 29.6 Conclusions ............................................ 29-23 29.1 Introduction Proteins play a key role in almost all biological processes. They take part in, for example, maintaining the structural integrity of the cell, transport and storage of small molecules, catalysis, regulation, signaling and the immune system. Linear protein molecules fold up into specific three-dimensional structures, and their functional properties depend intricately upon their structures. As a result, there has been much effort, both experimental and computational, in determining protein structures. Protein structures are determined experimentally using either x-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy. While -

Lecture 7 Protein Secondary Structure Prediction
Protein primary structure C Master Course E N DNA/Protein Structure- 20 amino acid types A generic residue Peptide bond T R E function Analysis and F B O I Prediction R O I I N N T F E O G R R M Lecture 7 SARS Protein From Staphylococcus Aureus A A 1 MKYNNHDKIR DFIIIEAYMF RFKKKVKPEV T T 31 DMTIKEFILL TYLFHQQENT LPFKKIVSDL I I 61 CYKQSDLVQH IKVLVKHSYI SKVRSKIDER V C 91 NTYISISEEQ REKIAERVTL FDQIIKQFNL E S 121 ADQSESQMIP KDSKEFLNLM MYTMYFKNII V Protein Secondary 151 KKHLTLSFVE FTILAIITSQ NKNIVLLKDL U 181 IETIHHKYPQ TVRALNNLKK QGYLIKERST 211 EDERKILIHM DDAQQDHAEQ LLAQVNQLLA Structure Prediction 241 DKDHLHLVFE Protein secondary structure Alpha-helix Beta strands/sheet Secondary Structure • An easier question – what is the secondary structure when the 3D structure is known? SARS Protein From Staphylococcus Aureus 1 MKYNNHDKIR DFIIIEAYMF RFKKKVKPEV DMTIKEFILL TYLFHQQENT SHHH HHHHHHHHHH HHHHHHTTT SS HHHHHHH HHHHS S SE 51 LPFKKIVSDL CYKQSDLVQH IKVLVKHSYI SKVRSKIDER NTYISISEEQ EEHHHHHHHS SS GGGTHHH HHHHHHTTS EEEE SSSTT EEEE HHH 101 REKIAERVTL FDQIIKQFNL ADQSESQMIP KDSKEFLNLM MYTMYFKNII HHHHHHHHHH HHHHHHHHHH HTT SS S SHHHHHHHH HHHHHHHHHH 151 KKHLTLSFVE FTILAIITSQ NKNIVLLKDL IETIHHKYPQ TVRALNNLKK HHH SS HHH HHHHHHHHTT TT EEHHHH HHHSSS HHH HHHHHHHHHH 201 QGYLIKERST EDERKILIHM DDAQQDHAEQ LLAQVNQLLA DKDHLHLVFE HTSSEEEE S SSTT EEEE HHHHHHHHH HHHHHHHHTS SS TT SS DSSP • DSSP (Dictionary of Secondary Structure of a Protein) – assigns secondary structure to proteins which have a crystal (x-ray) or NMR (Nuclear Magnetic Resonance) A more challenging task: structure Predicting secondary structure from H = alpha helix primary sequence alone B = beta bridge (isolated residue) DSSP uses hydrogen-bonding E = extended beta strand structure to assign Secondary Structure Elements (SSEs). -

The Development of the Prediction of Protein Structure
6 The Development of the Prediction of Protein Structure Gerald D. Fasman I. Introduction .................................................................... 194 II. Protein Topology. .. 196 III. Techniques of Protein Prediction ................................................... 198 A. Sequence Alignment .......................................................... 199 B. Hydrophobicity .............................................................. 200 C. Minimum Energy Calculations ................................................. 202 IV. Approaches to Protein Conformation ................................................ 203 A. Solvent Accessibility ......................................................... 203 B. Packing of Residues .......................................................... 204 C. Distance Geometry ........................................................... 205 D. Amino Acid Physicochemical Properties ......................................... 205 V. Prediction of the Secondary Structure of Proteins: a Helix, ~ Strands, and ~ Turn .......... 208 A. ~ Turns .................................................................... 209 B. Evaluation of Predictive Methodologies .......................................... 218 C. Other Predictive Algorithms ................................................... 222 D. Chou-Fasman Algorithm ...................................................... 224 E. Class Prediction ............................................................. 233 VI. Prediction of Tertiary Structure ...................................................