Lecture 7 Protein Secondary Structure Prediction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn
biomolecules Review The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn 1, 1, 1 2 1,3 Daniel Rademaker y, Jarek van Dijk y, Willem Titulaer , Joanna Lange , Gert Vriend and Li Xue 1,* 1 Centre for Molecular and Biomolecular Informatics (CMBI), Radboudumc, 6525 GA Nijmegen, The Netherlands; [email protected] (D.R.); [email protected] (J.v.D.); [email protected] (W.T.); [email protected] (G.V.) 2 Bio-Prodict, 6511 AA Nijmegen, The Netherlands; [email protected] 3 Baco Institute of Protein Science (BIPS), Mindoro 5201, Philippines * Correspondence: [email protected] These authors contributed equally to this work. y Received: 15 May 2020; Accepted: 2 June 2020; Published: 16 June 2020 Abstract: When Oleg Ptitsyn and his group published the first secondary structure prediction for a protein sequence, they started a research field that is still active today. Oleg Ptitsyn combined fundamental rules of physics with human understanding of protein structures. Most followers in this field, however, use machine learning methods and aim at the highest (average) percentage correctly predicted residues in a set of proteins that were not used to train the prediction method. We show that one single method is unlikely to predict the secondary structure of all protein sequences, with the exception, perhaps, of future deep learning methods based on very large neural networks, and we suggest that some concepts pioneered by Oleg Ptitsyn and his group in the 70s of the previous century likely are today’s best way forward in the protein secondary structure prediction field. -

Collagen and Creatine
COLLAGEN AND CREATINE : PROTEIN AND NONPROTEIN NITROGENOUS COMPOUNDS Color index: . Important . Extra explanation “ THERE IS NO ELEVATOR TO SUCCESS. YOU HAVE TO TAKE THE STAIRS ” 435 Biochemistry Team • Amino acid structure. • Proteins. • Level of protein structure. RECALL: 435 Biochemistry Team Amino acid structure 1- hydrogen atom *H* ( which is distictive for each amino 2- side chain *R* acid and gives the amino acid a unique set of characteristic ) - Carboxylic acid group *COOH* 3- two functional groups - Primary amino acid group *NH2* ( except for proline which has a secondary amino acid) .The amino acid with a free amino Group at the end called “N-Terminus” . Alpha carbon that is attached to: to: thatattachedAlpha carbon is .The amino acid with a free carboxylic group At the end called “ C-Terminus” Proteins Proteins structure : - Building blocks , made of small molecules unit called amino acid which attached together in long chain by a peptide bond . Level of protein structure Tertiary Quaternary Primary secondary Single amino acids Region stabilized by Three–dimensional attached by hydrogen bond between Association of covalent bonds atoms of the polypeptide (3D) shape of called peptide backbone. entire polypeptide multi polypeptides chain including forming a bonds to form a Examples : linear sequence of side chain (R functional protein. amino acids. Alpha helix group ) Beta sheet 435 Biochemistry Team Level of protein structure 435 Biochemistry Team Secondary structure Alpha helix: - It is right-handed spiral , which side chain extend outward. - it is stabilized by hydrogen bond , which is formed between the peptide bond carbonyl oxygen and amide hydrogen. - each turn contains 3.6 amino acids. -

Chapter 4 the Three-Dimensional Structure of Proteins
Chapter 4 The Three-Dimensional Structure of Proteins Multiple Choice Questions 1. Answer: D All of the following are considered “weak” interactions in proteins, except: A) hydrogen bonds. B) hydrophobic interactions. C) ionic bonds. D) peptide bonds. E) van der Waals forces. 2. Answer: D In an aqueous solution, protein conformation is determined by two major factors. One is the formation of the maximum number of hydrogen bonds. The other is the: A) formation of the maximum number of hydrophilic interactions. B) maximization of ionic interactions. C) minimization of entropy by the formation of a water solvent shell around the protein. D) placement of hydrophobic amino acid residues within the interior of the protein. E) placement of polar amino acid residues around the exterior of the protein. 3. 3 Answer: A In the diagram below, the plane drawn behind the peptide bond indicates the: A) absence of rotation around the C—N bond because of its partial double-bond character. B) plane of rotation around the C—N bond. C) region of steric hindrance determined by the large C=O group. D) region of the peptide bond that contributes to a Ramachandran plot. E) theoretical space between –180 and +180 degrees that can be occupied by the and angles in the peptide bond. 4. Answer: D Which of the following best represents the backbone arrangement of two peptide bonds? A) C—N—C—C—C—N—C—C B) C—N—C—C—N—C C) C—N—C—C—C—N D) C—C—N—C—C—N Chapter 4 The Three-Dimensional Structure of Proteins E) C—C—C—N—C—C—C 5. -

Conformational Properties of Constrained Proline Analogues and Their Application in Nanobiology”
UNIVERSITAT POLITÈCNICA DE CATALUNYA DEPARTAMENT D’ENGINYERIA QUÍMICA “CONFORMATIONAL PROPERTIES OF CONSTRAINED PROLINE ANALOGUES AND THEIR APPLICATION IN NANOBIOLOGY” Alejandra Flores Ortega Supervisors: Dr. Carlos Alemán Llansó and Dr. Jordi Casanovas Salas. th Barcelona, 27 January 2009 “Chance is a word void of sense; nothing can exist without a cause”. François-Marie Arouet, Voltaire “Imagination will often carry us to worlds that never were. But without it, we go nowhere”. Carl Sagan iii ACKNOWLEDGEMENTS I would like to acknowledge to Dr. Carlos Aleman and Dr. Jordi Cassanovas Salas for an interesting research theme, and scientific support. I gratefully acknowledge to Dr. David Zanuy for interesting suggestions and strong discussions, without their support this would be an unfulfilled task. Also I, would like to address my thanks to all my colleagues in my group and department, specially Elaine Armelin for assiting me in many different ways. I thank not only my friends, but also colleagues for helping me to overcome the stressful time, without whom it would have been difficult to cope up. I wish to express my gratefulness to my parents, specially to my mother, María Esther, for all his care, and support. Also I will like to thanks to my friends and specially Jesus, Merches, Laura y Arturo. My PhD thesis have been finished for all this support. I am greatly indepted to Dr. Ruth Nussinov at NCI, Dr. Carlos Cativiela at the University of Zaragoza and Ana I. Jiménez at the “Instituto de Ciencias de Materiales de Aragon” for a collaborative effort. I wish to thank all my colleague in the “Chimie et Biochimie Théoriques, Faculté des Sciences et Techniques” in Nancy France, I will be grateful to have worked with : Pr. -

2. Proteins Have Hierarchies of Structure
27 2. Proteins have Hierarchies of Structure Protein structure is usually described at four different levels (Fig. II.2.1). The first level, called the primary structure, describes the linear sequence of the amino acids in the chain. The different primary structures correspond to the different sequences in which the amino acids are covalently linked together. The secondary structure describes two common patterns of structural repetition in proteins: the coiling up into helices of segments of the chain, and the pairing together of strands of the chain into β-sheets. The tertiary structure is the next higher level of organization, the overall arrangement of secondary structural elements. The quaternary structure describes how different polypeptide chains are assembled into complexes. Figure II.2.1. Different levels of protein structure. A protein chain’s primary structure is its amino acid sequence. Secondary structure consists of the regular organization of helices and sheets. The example shown is a schematic representation of an α helix. Tertiary structure is a polypeptide chain’s three- dimensional native conformation, which often involves compact packing of secondary structure elements. The example given in the figure is a schematic drawing of one of the four polypeptide chains (subunits) of hemoglobin, the protein that transports oxygen in the blood. α-helices along the chain are represented as cylinders. N is the amino terminus and C is the carboxyl terminus of the polypeptide chain. Quaternary structure is the arrangement of multiple polypeptide chains (subunits) to form a functional biomolecular structure. The figure shows the quaternary arrangement of four subunits to form the functional hemoglobin molecule. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Detection of Trans-Cis Flips and Peptide-Plane Flips in Protein Structures
research papers Detection of trans–cis flips and peptide-plane flips in protein structures ISSN 1399-0047 Wouter G. Touw,a* Robbie P. Joostenb and Gert Vrienda* aCentre for Molecular and Biomolecular Informatics, Radboud University Medical Center, Geert Grooteplein-Zuid 26-28, 6525 GA Nijmegen, The Netherlands, and bDepartment of Biochemistry, Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. *Correspondence e-mail: [email protected], Received 4 February 2015 [email protected] Accepted 27 April 2015 A coordinate-based method is presented to detect peptide bonds that need Edited by G. J. Kleywegt, EMBL–EBI, Hinxton, correction either by a peptide-plane flip or by a trans–cis inversion of the peptide England bond. When applied to the whole Protein Data Bank, the method predicts 4617 trans–cis flips and many thousands of hitherto unknown peptide-plane flips. Keywords: peptide conformation; cis peptide A few examples are highlighted for which a correction of the peptide-plane bond; structure validation; structure correction. geometry leads to a correction of the understanding of the structure–function relation. All data, including 1088 manually validated cases, are freely available and the method is available from a web server, a web-service interface and through WHAT_CHECK. 1. Introduction Peptide bonds connect adjacent amino acids in proteins. The partial double-bond character of the peptide bond restricts its torsion. The dihedral angle ! (CiÀ1—CiÀ1—Ni—Ci ) typically has values around 180 (trans)or0 (cis), although exceptions are possible (Berkholz et al., 2012). The Ci–1—Ci distance is around 3.81 A˚ in the trans conformation and around 2.94 A˚ in the cis conformation. -

Packing of Secondary Structures II
7.88 Lecture Notes - 5 7.24/7.88J/5.48J The Protein Folding and Human Disease Packing of Secondary Structures • Packing of Helices against sheets • Packing of sheets against sheets • Parallel • Orthogonal Table: “Amino Acid Composition of the Ten Proteins and of the Residues at the Helix to Helix Interfaces” Name Total % Total At contacts % at contacts Gly 182 9 15 4 Ala 191 9 49 12 Val 151 7 46 12 Leu 148 7 48 12 Ile 114 6 36 9 Pro 67 3 41 1 Phe 68 3 25 6 Tyr 87 6 14 4 Trp 35 2 7 2 His 45 2 18 5 ½Cys 21 1 3 1 Met 29 1 10 3 Ser 165 8 19 5 Thr 132 7 21 5 Asp 112 6 14 4 Asn 113 6 13 3 Glu 94 5 13 3 Gln 70 3 12 3 Lys 125 6 19 5 Arg 76 4 13 3 A. Factors Contributing to Stability of Correctly Folded Native State 1. Major source of stability = removal of hydrophobic side chains atoms from the solvent and burying in environment which excludes the solvent (Entropic contribution from water structure). 1 2. Formation of hydrogen bonds between buried amide and carbonyl groups is maximized 3. Retention of backbone conformations close to the minimal energies. 4. Close packing means optimal Van der Waals interactions. You have read about alpha/beta proteins in Brandon and Tooze. B. Helix to Sheet Packing Lets examine buried contacts between the helices and the sheets. First a quick review of beta sheet structure: Colored transparency: Theoretical model, not actual sheet. -

Amino Acid Preference Against Beta Sheet Through Allowing Backbone Hydration Enabled by the Presence of Cation
Amino acid preference against beta sheet through allowing backbone hydration enabled by the presence of cation John N. Sharley, University of Adelaide. arXiv 2016-10-03 [email protected] Table of Contents 1 Abstract 1 2 Introduction 2 2.1 Alpha helix preferring amino acid residues in a beta sheet 2 2.2 Cation interactions with protein backbone oxygen 3 2.3 Quantum molecular dynamics with quantum mechanical treatment of every water molecule 3 3 Methods 4 4 Results 5 4.1 Preparation 5 4.2 Experiment 1302 5 4.3 Experiment 1303 7 5 Discussion 9 5.1 HB networks of water 9 5.2 Subsequent to rupture of a transient beta sheet 9 5.3 Hofmeister effects 10 6 Conclusion 11 7 Future work 12 8 Acknowledgements 13 9 References 14 10 Appendix 1. Backbone hydration in experiment 1302 16 11 Appendix 2. Backbone hydration in experiment 1303 17 1 Abstract It is known that steric blocking by peptide sidechains of hydrogen bonding, HB, between water and peptide groups, PGs, in beta sheets accords with an amino acid intrinsic beta sheet preference [1]. The present observations with Quantum Molecular Dynamics, QMD, simulation with Quantum Mechanical, QM, treatment of every water molecule solvating a beta sheet that would be transient in nature suggest that this steric blocking is not applicable in a hydrophobic region unless a cation is present, so that the amino acid beta sheet preference due to this steric blocking is only effective in the presence of a cation. We observed backbone hydration in a polyalanine and to a lesser extent polyvaline alpha helix without a cation being present, but a cation could increase the strength of these HBs. -
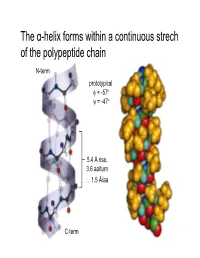
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Helix Stability of Oligoglycine, Oligoalanine, and Oligoalanine
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Helix stability of oligoglycine, oligoalanine, and oligo-b-alanine dodecamers reflected by hydrogen-bond persistence Chengyu Liu,1 Jay W. Ponder,1 and Garland R. Marshall2* 1 Department of Chemistry, Washington University, St. Louis, Missouri 63130 2 Department of Biochemistry and Molecular Biophysics, Washington University, St. Louis, Missouri 63130 ABSTRACT Helices are important structural/recognition elements in proteins and peptides. Stability and conformational differences between helices composed of a- and b-amino acids as scaffolds for mimicry of helix recognition has become a theme in medicinal chemistry. Furthermore, helices formed by b-amino acids are experimentally more stable than those formed by a-amino acids. This is paradoxical because the larger sizes of the hydrogen-bonding rings required by the extra methylene groups should lead to entropic destabilization. In this study, molecular dynamics simulations using the second-generation force field, AMOEBA (Ponder, J.W., et al., Current status of the AMOEBA polarizable force field. J Phys Chem B, 2010. 114(8): p. 2549–64.) explored the stability and hydrogen-bonding patterns of capped oligo-b-alanine, oligoalanine, and oligo- glycine dodecamers in water. The MD simulations showed that oligo-b-alanine has strong acceptor12 hydrogen bonds, but surprisingly did not contain a large content of 312-helical structures, possibly due to the sparse distribution of the 312-helical structure and other structures with acceptor12 hydrogen bonds. On the other hand, despite its backbone flexibility, the b- alanine dodecamer had more stable and persistent <3.0 A˚ hydrogen bonds. Its structure was dominated more by multicen- tered hydrogen bonds than either oligoglycine or oligoalanine helices. -

Helix Capping'
Prorein Science (1998), 721-38. Cambridge University Press. Printed in the USA. Copyright 0 1998 The Protein Society REVIEW Helix capping' RAJEEV AURORA AND GEORGE D. ROSE Department of Biophysics and Biophysical Chemistry, Johns Hopkins University School of Medicine, 725 N. Wolfe Street, Baltimore, Maryland 21205 (RECEIVED June12, 1997; ACCEPTEDJuly 9, 1997) Abstract Helix-capping motifs are specific patterns of hydrogen bonding and hydrophobic interactions found at or near the ends of helices in both proteins and peptides. In an a-helix, the first four >N- H groups and last four >C=O groups necessarily lack intrahelical hydrogen bonds. Instead, such groups are often capped by alternative hydrogen bond partners. This review enlarges our earlier hypothesis (Presta LG, Rose GD. 1988. Helix signals in proteins. Science 240:1632-1641) to include hydrophobic capping. A hydrophobic interaction that straddles the helix terminus is always associated with hydrogen-bonded capping. From a global survey among proteins of known structure, seven distinct capping motifs are identified-three at the helix N-terminus and four at the C-terminus. The consensus sequence patterns of these seven motifs, together with results from simple molecular modeling, are used to formulate useful rules of thumb for helix termination. Finally, we examine the role of helix capping as a bridge linking the conformation of secondary structure to supersecondary structure. Keywords: alpha helix; protein folding; protein secondary structure The a-helixis characterized by consecutive, main-chain, i + i - 4 apolar residues in the a-helix and its flanking turn. This hydro- hydrogen bonds between each amide hydrogen and a carbonyl phobic component of helix capping was unanticipated.