PA28: New Insights on an Ancient Proteasome Activator
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn
biomolecules Review The Future of Protein Secondary Structure Prediction Was Invented by Oleg Ptitsyn 1, 1, 1 2 1,3 Daniel Rademaker y, Jarek van Dijk y, Willem Titulaer , Joanna Lange , Gert Vriend and Li Xue 1,* 1 Centre for Molecular and Biomolecular Informatics (CMBI), Radboudumc, 6525 GA Nijmegen, The Netherlands; [email protected] (D.R.); [email protected] (J.v.D.); [email protected] (W.T.); [email protected] (G.V.) 2 Bio-Prodict, 6511 AA Nijmegen, The Netherlands; [email protected] 3 Baco Institute of Protein Science (BIPS), Mindoro 5201, Philippines * Correspondence: [email protected] These authors contributed equally to this work. y Received: 15 May 2020; Accepted: 2 June 2020; Published: 16 June 2020 Abstract: When Oleg Ptitsyn and his group published the first secondary structure prediction for a protein sequence, they started a research field that is still active today. Oleg Ptitsyn combined fundamental rules of physics with human understanding of protein structures. Most followers in this field, however, use machine learning methods and aim at the highest (average) percentage correctly predicted residues in a set of proteins that were not used to train the prediction method. We show that one single method is unlikely to predict the secondary structure of all protein sequences, with the exception, perhaps, of future deep learning methods based on very large neural networks, and we suggest that some concepts pioneered by Oleg Ptitsyn and his group in the 70s of the previous century likely are today’s best way forward in the protein secondary structure prediction field. -

Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins
International Journal of Molecular Sciences Article Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins M. Quadir Siddiqui 1,† , Maulik D. Badmalia 1,† and Trushar R. Patel 1,2,3,* 1 Alberta RNA Research and Training Institute, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive, Lethbridge, AB T1K 3M4, Canada; [email protected] (M.Q.S.); [email protected] (M.D.B.) 2 Department of Microbiology, Immunology and Infectious Disease, Cumming School of Medicine, University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada 3 Li Ka Shing Institute of Virology, University of Alberta, Edmonton, AB T6G 2E1, Canada * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Members of the human Zyxin family are LIM domain-containing proteins that perform critical cellular functions and are indispensable for cellular integrity. Despite their importance, not much is known about their structure, functions, interactions and dynamics. To provide insights into these, we used a set of in-silico tools and databases and analyzed their amino acid sequence, phylogeny, post-translational modifications, structure-dynamics, molecular interactions, and func- tions. Our analysis revealed that zyxin members are ohnologs. Presence of a conserved nuclear export signal composed of LxxLxL/LxxxLxL consensus sequence, as well as a possible nuclear localization signal, suggesting that Zyxin family members may have nuclear and cytoplasmic roles. The molecular modeling and structural analysis indicated that Zyxin family LIM domains share Citation: Siddiqui, M.Q.; Badmalia, similarities with transcriptional regulators and have positively charged electrostatic patches, which M.D.; Patel, T.R. -

Phylogenomic Analysis of the Chlamydomonas Genome Unmasks Proteins Potentially Involved in Photosynthetic Function and Regulation
Photosynth Res DOI 10.1007/s11120-010-9555-7 REVIEW Phylogenomic analysis of the Chlamydomonas genome unmasks proteins potentially involved in photosynthetic function and regulation Arthur R. Grossman • Steven J. Karpowicz • Mark Heinnickel • David Dewez • Blaise Hamel • Rachel Dent • Krishna K. Niyogi • Xenie Johnson • Jean Alric • Francis-Andre´ Wollman • Huiying Li • Sabeeha S. Merchant Received: 11 February 2010 / Accepted: 16 April 2010 Ó The Author(s) 2010. This article is published with open access at Springerlink.com Abstract Chlamydomonas reinhardtii, a unicellular green performed to identify proteins encoded on the Chlamydo- alga, has been exploited as a reference organism for iden- monas genome which were likely involved in chloroplast tifying proteins and activities associated with the photo- functions (or specifically associated with the green algal synthetic apparatus and the functioning of chloroplasts. lineage); this set of proteins has been designated the Recently, the full genome sequence of Chlamydomonas GreenCut. Further analyses of those GreenCut proteins with was generated and a set of gene models, representing all uncharacterized functions and the generation of mutant genes on the genome, was developed. Using these gene strains aberrant for these proteins are beginning to unmask models, and gene models developed for the genomes of new layers of functionality/regulation that are integrated other organisms, a phylogenomic, comparative analysis was into the workings of the photosynthetic apparatus. Keywords Chlamydomonas Á GreenCut Á Chloroplast Á Phylogenomics Á Regulation A. R. Grossman (&) Á M. Heinnickel Á D. Dewez Á B. Hamel Department of Plant Biology, Carnegie Institution for Science, 260 Panama Street, Stanford, CA 94305, USA Introduction e-mail: [email protected] Chlamydomonas reinhardtii as a reference organism S. -

An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale † ‡ ‡ ‡ § ‡ ∥ Upneet Kaur, He Meng, Fang Lui, Renze Ma, Ryenne N
Perspective Cite This: J. Proteome Res. 2018, 17, 3614−3627 pubs.acs.org/jpr Proteome-Wide Structural Biology: An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale † ‡ ‡ ‡ § ‡ ∥ Upneet Kaur, He Meng, Fang Lui, Renze Ma, Ryenne N. Ogburn, , Julia H. R. Johnson, , ‡ † Michael C. Fitzgerald,*, and Lisa M. Jones*, ‡ Department of Chemistry, Duke University, Durham, North Carolina 27708-0346, United States † Department of Pharmaceutical Sciences, University of Maryland, Baltimore, Maryland 21201, United States ABSTRACT: Over the past decade, a suite of new mass- spectrometry-based proteomics methods has been developed that now enables the conformational properties of proteins and protein− ligand complexes to be studied in complex biological mixtures, from cell lysates to intact cells. Highlighted here are seven of the techniques in this new toolbox. These techniques include chemical cross-linking (XL−MS), hydroxyl radical footprinting (HRF), Drug Affinity Responsive Target Stability (DARTS), Limited Proteolysis (LiP), Pulse Proteolysis (PP), Stability of Proteins from Rates of Oxidation (SPROX), and Thermal Proteome Profiling (TPP). The above techniques all rely on conventional bottom-up proteomics strategies for peptide sequencing and protein identification. However, they have required the development of unconventional proteomic data analysis strategies. Discussed here are the current technical challenges associated with these different data analysis strategies as well as the relative analytical capabilities of the different techniques. The new biophysical capabilities that the above techniques bring to bear on proteomic research are also highlighted in the context of several different application areas in which these techniques have been used, including the study of protein ligand binding interactions (e.g., protein target discovery studies and protein interaction network analyses) and the characterization of biological states. -

Chapter 4 the Three-Dimensional Structure of Proteins
Chapter 4 The Three-Dimensional Structure of Proteins Multiple Choice Questions 1. Answer: D All of the following are considered “weak” interactions in proteins, except: A) hydrogen bonds. B) hydrophobic interactions. C) ionic bonds. D) peptide bonds. E) van der Waals forces. 2. Answer: D In an aqueous solution, protein conformation is determined by two major factors. One is the formation of the maximum number of hydrogen bonds. The other is the: A) formation of the maximum number of hydrophilic interactions. B) maximization of ionic interactions. C) minimization of entropy by the formation of a water solvent shell around the protein. D) placement of hydrophobic amino acid residues within the interior of the protein. E) placement of polar amino acid residues around the exterior of the protein. 3. 3 Answer: A In the diagram below, the plane drawn behind the peptide bond indicates the: A) absence of rotation around the C—N bond because of its partial double-bond character. B) plane of rotation around the C—N bond. C) region of steric hindrance determined by the large C=O group. D) region of the peptide bond that contributes to a Ramachandran plot. E) theoretical space between –180 and +180 degrees that can be occupied by the and angles in the peptide bond. 4. Answer: D Which of the following best represents the backbone arrangement of two peptide bonds? A) C—N—C—C—C—N—C—C B) C—N—C—C—N—C C) C—N—C—C—C—N D) C—C—N—C—C—N Chapter 4 The Three-Dimensional Structure of Proteins E) C—C—C—N—C—C—C 5. -

Conformational Properties of Constrained Proline Analogues and Their Application in Nanobiology”
UNIVERSITAT POLITÈCNICA DE CATALUNYA DEPARTAMENT D’ENGINYERIA QUÍMICA “CONFORMATIONAL PROPERTIES OF CONSTRAINED PROLINE ANALOGUES AND THEIR APPLICATION IN NANOBIOLOGY” Alejandra Flores Ortega Supervisors: Dr. Carlos Alemán Llansó and Dr. Jordi Casanovas Salas. th Barcelona, 27 January 2009 “Chance is a word void of sense; nothing can exist without a cause”. François-Marie Arouet, Voltaire “Imagination will often carry us to worlds that never were. But without it, we go nowhere”. Carl Sagan iii ACKNOWLEDGEMENTS I would like to acknowledge to Dr. Carlos Aleman and Dr. Jordi Cassanovas Salas for an interesting research theme, and scientific support. I gratefully acknowledge to Dr. David Zanuy for interesting suggestions and strong discussions, without their support this would be an unfulfilled task. Also I, would like to address my thanks to all my colleagues in my group and department, specially Elaine Armelin for assiting me in many different ways. I thank not only my friends, but also colleagues for helping me to overcome the stressful time, without whom it would have been difficult to cope up. I wish to express my gratefulness to my parents, specially to my mother, María Esther, for all his care, and support. Also I will like to thanks to my friends and specially Jesus, Merches, Laura y Arturo. My PhD thesis have been finished for all this support. I am greatly indepted to Dr. Ruth Nussinov at NCI, Dr. Carlos Cativiela at the University of Zaragoza and Ana I. Jiménez at the “Instituto de Ciencias de Materiales de Aragon” for a collaborative effort. I wish to thank all my colleague in the “Chimie et Biochimie Théoriques, Faculté des Sciences et Techniques” in Nancy France, I will be grateful to have worked with : Pr. -

2. Proteins Have Hierarchies of Structure
27 2. Proteins have Hierarchies of Structure Protein structure is usually described at four different levels (Fig. II.2.1). The first level, called the primary structure, describes the linear sequence of the amino acids in the chain. The different primary structures correspond to the different sequences in which the amino acids are covalently linked together. The secondary structure describes two common patterns of structural repetition in proteins: the coiling up into helices of segments of the chain, and the pairing together of strands of the chain into β-sheets. The tertiary structure is the next higher level of organization, the overall arrangement of secondary structural elements. The quaternary structure describes how different polypeptide chains are assembled into complexes. Figure II.2.1. Different levels of protein structure. A protein chain’s primary structure is its amino acid sequence. Secondary structure consists of the regular organization of helices and sheets. The example shown is a schematic representation of an α helix. Tertiary structure is a polypeptide chain’s three- dimensional native conformation, which often involves compact packing of secondary structure elements. The example given in the figure is a schematic drawing of one of the four polypeptide chains (subunits) of hemoglobin, the protein that transports oxygen in the blood. α-helices along the chain are represented as cylinders. N is the amino terminus and C is the carboxyl terminus of the polypeptide chain. Quaternary structure is the arrangement of multiple polypeptide chains (subunits) to form a functional biomolecular structure. The figure shows the quaternary arrangement of four subunits to form the functional hemoglobin molecule. -

Introduction to Proteins and Amino Acids Introduction
Introduction to Proteins and Amino Acids Introduction • Twenty percent of the human body is made up of proteins. Proteins are the large, complex molecules that are critical for normal functioning of cells. • They are essential for the structure, function, and regulation of the body’s tissues and organs. • Proteins are made up of smaller units called amino acids, which are building blocks of proteins. They are attached to one another by peptide bonds forming a long chain of proteins. Amino acid structure and its classification • An amino acid contains both a carboxylic group and an amino group. Amino acids that have an amino group bonded directly to the alpha-carbon are referred to as alpha amino acids. • Every alpha amino acid has a carbon atom, called an alpha carbon, Cα ; bonded to a carboxylic acid, –COOH group; an amino, –NH2 group; a hydrogen atom; and an R group that is unique for every amino acid. Classification of amino acids • There are 20 amino acids. Based on the nature of their ‘R’ group, they are classified based on their polarity as: Classification based on essentiality: Essential amino acids are the amino acids which you need through your diet because your body cannot make them. Whereas non essential amino acids are the amino acids which are not an essential part of your diet because they can be synthesized by your body. Essential Non essential Histidine Alanine Isoleucine Arginine Leucine Aspargine Methionine Aspartate Phenyl alanine Cystine Threonine Glutamic acid Tryptophan Glycine Valine Ornithine Proline Serine Tyrosine Peptide bonds • Amino acids are linked together by ‘amide groups’ called peptide bonds. -

Detection of Trans-Cis Flips and Peptide-Plane Flips in Protein Structures
research papers Detection of trans–cis flips and peptide-plane flips in protein structures ISSN 1399-0047 Wouter G. Touw,a* Robbie P. Joostenb and Gert Vrienda* aCentre for Molecular and Biomolecular Informatics, Radboud University Medical Center, Geert Grooteplein-Zuid 26-28, 6525 GA Nijmegen, The Netherlands, and bDepartment of Biochemistry, Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. *Correspondence e-mail: [email protected], Received 4 February 2015 [email protected] Accepted 27 April 2015 A coordinate-based method is presented to detect peptide bonds that need Edited by G. J. Kleywegt, EMBL–EBI, Hinxton, correction either by a peptide-plane flip or by a trans–cis inversion of the peptide England bond. When applied to the whole Protein Data Bank, the method predicts 4617 trans–cis flips and many thousands of hitherto unknown peptide-plane flips. Keywords: peptide conformation; cis peptide A few examples are highlighted for which a correction of the peptide-plane bond; structure validation; structure correction. geometry leads to a correction of the understanding of the structure–function relation. All data, including 1088 manually validated cases, are freely available and the method is available from a web server, a web-service interface and through WHAT_CHECK. 1. Introduction Peptide bonds connect adjacent amino acids in proteins. The partial double-bond character of the peptide bond restricts its torsion. The dihedral angle ! (CiÀ1—CiÀ1—Ni—Ci ) typically has values around 180 (trans)or0 (cis), although exceptions are possible (Berkholz et al., 2012). The Ci–1—Ci distance is around 3.81 A˚ in the trans conformation and around 2.94 A˚ in the cis conformation. -

Static Retention of the Lumenal Monotopic Membrane Protein Torsina in the Endoplasmic Reticulum
The EMBO Journal (2011) 30, 3217–3231 | & 2011 European Molecular Biology Organization | All Rights Reserved 0261-4189/11 www.embojournal.org TTHEH E EEMBOMBO JJOURNALOURN AL Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum Abigail B Vander Heyden1, despite the fact that it has been a decade since the protein was Teresa V Naismith1, Erik L Snapp2 and first described and linked to dystonia (Breakefield et al, Phyllis I Hanson1,* 2008). Based on its membership in the AAA þ family of ATPases (Ozelius et al, 1997; Hanson and Whiteheart, 2005), 1Department of Cell Biology and Physiology, Washington University School of Medicine, St Louis, MO, USA and 2Department of Anatomy it is likely that torsinA disassembles or changes the confor- and Structural Biology, Albert Einstein College of Medicine, Bronx, mation of a protein or protein complex in the ER or NE. The NY, USA DE mutation is thought to compromise this function (Dang et al, 2005; Goodchild et al, 2005). TorsinA is a membrane-associated enzyme in the endo- TorsinA is targeted to the ER lumen by an N-terminal plasmic reticulum (ER) lumen that is mutated in DYT1 signal peptide. Analyses of torsinA’s subcellular localization, dystonia. How it remains in the ER has been unclear. We processing, and glycosylation show that the signal peptide is report that a hydrophobic N-terminal domain (NTD) di- cleaved and the mature protein resides in the lumen of the ER rects static retention of torsinA within the ER by excluding (Kustedjo et al, 2000; Hewett et al, 2003; Liu et al, 2003), it from ER exit sites, as has been previously reported for where it is a stable protein (Gordon and Gonzalez-Alegre, short transmembrane domains (TMDs). -
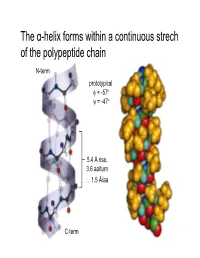
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

PROTEOMICS the Human Proteome Takes the Spotlight
RESEARCH HIGHLIGHTS PROTEOMICS The human proteome takes the spotlight Two papers report mass spectrometry– big data. “We then thought, include some surpris- based draft maps of the human proteome ‘What is a potentially good ing findings. For example, and provide broadly accessible resources. illustration for the utility Kuster’s team found protein For years, members of the proteomics of such a database?’” says evidence for 430 long inter- community have been trying to garner sup- Kuster. “We very quickly genic noncoding RNAs, port for a large-scale project to exhaustively got to the idea, ‘Why don’t which have been thought map the normal human proteome, including we try to put together the not to be translated into pro- identifying all post-translational modifica- human proteome?’” tein. Pandey’s team refined tions and protein-protein interactions and The two groups took the annotations of 808 genes providing targeted mass spectrometry assays slightly different strategies and also found evidence and antibodies for all human proteins. But a towards this common goal. for the translation of many Nik Spencer/Nature Publishing Group Publishing Nik Spencer/Nature lack of consensus on how to exactly define Pandey’s lab examined 30 noncoding RNAs and pseu- Two groups provide mass the proteome, how to carry out such a mis- normal tissues, including spectrometry evidence for dogenes. sion and whether the technology is ready has adult and fetal tissues, as ~90% of the human proteome. Obtaining evidence for not so far convinced any funding agencies to well as primary hematopoi- the last roughly 10% of pro- fund on such an ambitious project.