Primepcr™Assay Validation Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

DNA Methylation Changes in Down Syndrome Derived Neural Ipscs Uncover Co-Dysregulation of ZNF and HOX3 Families of Transcription
Laan et al. Clinical Epigenetics (2020) 12:9 https://doi.org/10.1186/s13148-019-0803-1 RESEARCH Open Access DNA methylation changes in Down syndrome derived neural iPSCs uncover co- dysregulation of ZNF and HOX3 families of transcription factors Loora Laan1†, Joakim Klar1†, Maria Sobol1, Jan Hoeber1, Mansoureh Shahsavani2, Malin Kele2, Ambrin Fatima1, Muhammad Zakaria1, Göran Annerén1, Anna Falk2, Jens Schuster1 and Niklas Dahl1* Abstract Background: Down syndrome (DS) is characterized by neurodevelopmental abnormalities caused by partial or complete trisomy of human chromosome 21 (T21). Analysis of Down syndrome brain specimens has shown global epigenetic and transcriptional changes but their interplay during early neurogenesis remains largely unknown. We differentiated induced pluripotent stem cells (iPSCs) established from two DS patients with complete T21 and matched euploid donors into two distinct neural stages corresponding to early- and mid-gestational ages. Results: Using the Illumina Infinium 450K array, we assessed the DNA methylation pattern of known CpG regions and promoters across the genome in trisomic neural iPSC derivatives, and we identified a total of 500 stably and differentially methylated CpGs that were annotated to CpG islands of 151 genes. The genes were enriched within the DNA binding category, uncovering 37 factors of importance for transcriptional regulation and chromatin structure. In particular, we observed regional epigenetic changes of the transcription factor genes ZNF69, ZNF700 and ZNF763 as well as the HOXA3, HOXB3 and HOXD3 genes. A similar clustering of differential methylation was found in the CpG islands of the HIST1 genes suggesting effects on chromatin remodeling. Conclusions: The study shows that early established differential methylation in neural iPSC derivatives with T21 are associated with a set of genes relevant for DS brain development, providing a novel framework for further studies on epigenetic changes and transcriptional dysregulation during T21 neurogenesis. -
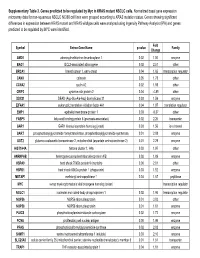
Supplementary Table 3. Genes Predicted to Be Regulated by Myc in KRAS Mutant NSCLC Cells
Supplementary Table 3. Genes predicted to be regulated by Myc in KRAS mutant NSCLC cells. Normalized basal gene expression microarray data for non-squamous NSCLC NCI60 cell lines were grouped according to KRAS mutation status. Genes showing significant differences in expression between KRAS mutant and KRAS wildtype cells were analyzed using Ingenuity Pathway Analysis (IPA) and genes predicted to be regulated by MYC were identified. Fold Symbol Entrez Gene Name p-value Family Change AMD1 adenosylmethionine decarboxylase 1 0.02 1.50 enzyme BAG1 BCL2-associated athanogene 0.02 2.57 other BRCA1 breast cancer 1, early onset 0.04 1.65 transcription regulator CANX calnexin 0.05 1.78 other CCNA2 cyclin A2 0.02 1.98 other CRIP2 cysteine-rich protein 2 0.04 -4.89 other DDX21 DEAD (Asp-Glu-Ala-Asp) box helicase 21 0.03 1.59 enzyme EIF4A1 eukaryotic translation initiation factor 4A1 0.04 1.87 translation regulator EMP1 epithelial membrane protein 1 0.03 -6.37 other FABP5 fatty acid binding protein 5 (psoriasis-associated) 0.02 2.26 transporter GAR1 GAR1 ribonucleoprotein homolog (yeast) 0.03 1.56 ion channel GART phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, 0.01 2.08 enzyme GOT2 glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) 0.01 2.29 enzyme HIST1H4A histone cluster 1, H4a 0.03 1.97 other HNRNPAB heterogeneous nuclear ribonucleoprotein A/B 0.02 1.89 enzyme HSPA9 heat shock 70kDa protein 9 (mortalin) 0.00 2.01 other HSPD1 heat shock 60kDa protein 1 (chaperonin) 0.03 1.52 enzyme -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -

Human Recombinant Protein – TP760406
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP760406 H4C1 (NM_003538) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Purified recombinant protein of Human histone cluster 1, H4a (HIST1H4A), full length, with N- terminal HIS tag, expressed in E.Coli, 50ug Species: Human Expression Host: E. coli Tag: N-His Predicted MW: 11.2 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25mM Tris, pH8.0, 150 mM NaCl, 10% glycerol, 1 % Sarkosyl Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_003529 Locus ID: 8359 UniProt ID: P62805, B2R4R0 RefSeq Size: 372 Cytogenetics: 6p22.2 RefSeq ORF: 309 Synonyms: H4-16; H4C2; H4C3; H4C4; H4C5; H4C6; H4C8; H4C9; H4C11; H4C12; H4C13; H4C14; H4C15; H4FA; HIST1H4A This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 H4C1 (NM_003538) Human Recombinant Protein – TP760406 Summary: Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A, H2B, H3, and H4) form an octamer, around which approximately 146 bp of DNA is wrapped in repeating units, called nucleosomes. -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

A Network Based Approach to Identify the Genetic Influence Caused By
bioRxiv preprint doi: https://doi.org/10.1101/482760; this version posted November 30, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Network based Approach to Identify the Genetic Influence Caused by Associated Factors and Disorders for the Parkinsons Disease Progression 1st Najmus Sakib 1st Utpala Nanda Chowdhury Dept. of Applied Physics and Electronic Engineering Dept. of Computer Science and Engineering University of Rajshahi University of Rajshahi Rakshahi, Bangladesh Rakshahi, Bangladesh najmus:sakib1995@outlook:com unchowdhury@ru:ac:bd 2nd M. Babul Islam 3rd Julian M.W. Quinn 4th Mohammad Ali Moni Dept. of Applied Physics and Electronic Engg. Bone biology divisions School of Medical Science University of Rajshahi Garvan Institute of Medical Research The University of Sydney Rakshahi, Bangladesh NSW 2010 NSW, Australia babul:apee@ru:ac:bd j:quinn@garvan:org:au mohammad:moni@sydney:edu:au Abstract—Actual causes of Parkinsons disease (PD) are still the central nervous system [1]. It is one of the most common unknown. In any case, a better comprehension of genetic and neurodegenerative problems after Alzheimers illness all over ecological influences to the PD and their interaction will assist the world [2]. The PD is characterized by progressive damage physicians and patients to evaluate individual hazard for the PD, and definitely, there will be a possibility to find a way to of dopaminergic neurons in the substantia nigra pars compacta reduce the progression of the PD. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Direct Antiviral Treatments for Hepatitis C Virus Have Off-Target
cancers Article Direct Antiviral Treatments for Hepatitis C Virus Have Off-Target Effects of Oncologic Relevance in Hepatocellular Carcinoma Catia Giovannini 1,2 , Francesca Fornari 1,2,* , Valentina Indio 3 , Davide Trerè 4 , Matteo Renzulli 5 , Francesco Vasuri 6, Matteo Cescon 2,7, Matteo Ravaioli 7, Alessia Perrucci 1, Annalisa Astolfi 3 , Fabio Piscaglia 2,8 and Laura Gramantieri 1,8,* 1 Center for Applied Biomedical Research (CRBA), Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy; [email protected] (C.G.); [email protected] (A.P.) 2 Department of Medical and Surgical Sciences, University of Bologna, 40138 Bologna, Italy; [email protected] (M.C.); [email protected] (F.P.) 3 “Giorgio Prodi” Cancer Research Center (CIRC), University of Bologna, 40138 Bologna, Italy; [email protected] (V.I.); annalisa.astolfi@unibo.it (A.A.) 4 Program of Laboratory Medicine, Azienda Ospedaliero-Universitaria di Bologna and Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, 40138 Bologna, Italy; [email protected] 5 Radiology Unit, Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy; [email protected] 6 Pathology Unit, Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy; [email protected] 7 Department of Surgery, Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy; [email protected] 8 Division of Internal Medicine Unit, Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy -

A Multiprotein Occupancy Map of the Mrnp on the 3 End of Histone
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press A multiprotein occupancy map of the mRNP on the 3′ end of histone mRNAs LIONEL BROOKS III,1 SHAWN M. LYONS,2 J. MATTHEW MAHONEY,1 JOSHUA D. WELCH,3 ZHONGLE LIU,1 WILLIAM F. MARZLUFF,2 and MICHAEL L. WHITFIELD1 1Department of Genetics, Dartmouth Geisel School of Medicine, Hanover, New Hampshire 03755, USA 2Integrative Program for Biological and Genome Sciences, University of North Carolina, Chapel Hill, North Carolina 27599, USA 3Department of Computer Science, University of North Carolina, Chapel Hill, North Carolina 27599, USA ABSTRACT The animal replication-dependent (RD) histone mRNAs are coordinately regulated with chromosome replication. The RD-histone mRNAs are the only known cellular mRNAs that are not polyadenylated. Instead, the mature transcripts end in a conserved stem– loop (SL) structure. This SL structure interacts with the stem–loop binding protein (SLBP), which is involved in all aspects of RD- histone mRNA metabolism. We used several genomic methods, including high-throughput sequencing of cross-linked immunoprecipitate (HITS-CLIP) to analyze the RNA-binding landscape of SLBP. SLBP was not bound to any RNAs other than histone mRNAs. We performed bioinformatic analyses of the HITS-CLIP data that included (i) clustering genes by sequencing read coverage using CVCA, (ii) mapping the bound RNA fragment termini, and (iii) mapping cross-linking induced mutation sites (CIMS) using CLIP-PyL software. These analyses allowed us to identify specific sites of molecular contact between SLBP and its RD-histone mRNA ligands. We performed in vitro crosslinking assays to refine the CIMS mapping and found that uracils one and three in the loop of the histone mRNA SL preferentially crosslink to SLBP, whereas uracil two in the loop preferentially crosslinks to a separate component, likely the 3′hExo. -

Exosomes Involved in Cholesterol Metabolism Process of Patients with Spinal Cord Injury in the Acute Phase
ORIGINAL RESEARCH published: 09 July 2021 doi: 10.3389/fninf.2021.662967 Bioinformatic Analysis of the Proteome in Exosomes Derived From Plasma: Exosomes Involved in Cholesterol Metabolism Process of Patients With Spinal Cord Injury in the Acute Phase Chunshuai Wu 1†, Jinjuan Yu 2†, Guanhua Xu 1, Hong Gao 1, Yue Sun 1, Jiayi Huang 1, Li Sun 1, Xu Zhang 1 and Zhiming Cui 1* 1 Department of Spine Surgery, Nantong First People’s Hospital, The Affiliated Hospital 2 of Nantong University, Nantong, China, 2 Department of Administrative Office, The Third People’s Hospital of Nantong, Nantong, China Edited by: Sandeep Kumar Dhanda, St. Jude Children’s Research Hospital, Spinal cord injury (SCI) is a common but severe disease caused by traffic accidents. United States Coronary atherosclerotic heart disease (CHD) caused by dyslipidemia is known as the Reviewed by: leading cause of death in patients with SCI. However, the quantitative analysis showed Prashanth N. Suravajhala, Birla Institute of Scientific that the cholesterol and lipoprotein concentrations in peripheral blood (PB) did not Research, India change significantly within 48 h after SCI. Due to the presence of the Blood spinal Chandrabose Selvaraj, Alagappa University, India cord barrier (BSCB), there are only few studies concerning the plasma cholesterol Aditya Ambati, metabolism in the acute phase of SCI. Exosomes have a smaller particle size, which Stanford University, United States enables them relatively less limitation of BSCB. This study uses exosomes derived from *Correspondence: the plasma of 43 patients in the acute phase of SCI and 71 patients in the control Zhiming Cui [email protected] group as samples. -

Research Article Common Expression Quantitative Trait Loci Shared by Histone Genes
Hindawi International Journal of Genomics Volume 2017, Article ID 6202567, 14 pages https://doi.org/10.1155/2017/6202567 Research Article Common Expression Quantitative Trait Loci Shared by Histone Genes Hanseol Kim, Yujin Suh, and Chaeyoung Lee Department of Bioinformatics and Life Science, Soongsil University, Seoul, Republic of Korea Correspondence should be addressed to Chaeyoung Lee; [email protected] Received 29 May 2017; Revised 26 July 2017; Accepted 2 August 2017; Published 27 August 2017 Academic Editor: Marco Gerdol Copyright © 2017 Hanseol Kim et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. A genome-wide association study (GWAS) was conducted to examine expression quantitative trait loci (eQTLs) for histone genes. We examined common eQTLs for multiple histone genes in 373 European lymphoblastoid cell lines (LCLs). A linear regression model was employed to identify single-nucleotide polymorphisms (SNPs) associated with expression of the histone genes, and the number of eQTLs was determined by linkage disequilibrium analysis. Additional associations of the identified eQTLs with other genes were also examined. We identified 31 eQTLs for 29 histone genes through genome-wide analysis using 29 histone genes (P <297 × 10−10). Among them, 12 eQTLs were associated with the expression of multiple histone genes. Transcriptome- wide association analysis using the identified eQTLs showed their associations with additional 80 genes (P <475 × 10−6). In particular, expression of RPPH1, SCARNA2, and SCARNA7 genes was associated with 26, 25, and 23 eQTLs, respectively.