In Silico Edgetic Profiling and Network Analysis of Human Genetic Variants, with an Application to Disease Module Detection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Sociology 342-001: Criminology Summer II
Sociology 342-001: Criminology Summer II: July 8 – Aug. 7 2013 Online - 3 credits Instructor Office Hours Kate Gunby via email and gchat [email protected] or by appointment in Social Sciences 426 Course Description This course begins with a quick introduction to the multidisciplinary study of criminology, and how crime and criminal behavior are measured. Then the class will explore different theories of crime and criminality, starting with early schools of criminology and then covering structural, social process, critical, psychosocial, biosocial, and developmental theories. Then the class will focus on different types of crime, including violent crime, sex crimes, multiple murder and terrorism, property crime, public order crime, and white collar and organized crime. Finally, we will broaden our scope to explore victim experiences, mental health and incarceration, concepts of justice and incarceration trends, and the consequences of crime and incarceration. This course uses the acclaimed television series The Wire to explore the fundamentals of criminology. Students will develop their ability analyze, synthesize, apply, and evaluate the course material through written memos linking each reading to the content in a specific episode of The Wire. Students will further engage with the material and each other through online forum discussions. This class is guided by student goals, which are established from the beginning and reviewed throughout the term. Readings All of the course readings are on D2L. You do not need to buy any books. Almost all of the readings are excerpts from books or articles, so please download the readings from D2L so that you only read the portions that are required for the class. -

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

The Wire: a Comprehensive List of Resources
The Wire: A comprehensive list of resources Contents Introduction ............................................................................................................................ 2 W: Academic Work on The Wire........................................................................................... 3 G: General Academic Work ................................................................................................... 9 I: Wire Related Internet Sources .......................................................................................... 11 1 Introduction William Julius Wilson has argued that: "The Wire’s exploration of sociological themes is truly exceptional. Indeed I do not hesitate to say that it has done more to enhance our understandings of the challenges of urban life and urban inequality than any other media event or scholarly publication, including studies by social scientists…The Wire develops morally complex characters on each side of the law, and with its scrupulous exploration of the inner workings of various institutions, including drug-dealing gangs, the police, politicians, unions, public schools, and the print media, viewers become aware that individuals’ decisions and behaviour are often shaped by - and indeed limited by - social, political, and economic forces beyond their control". Professor William Julius Wilson, Harvard University Seminar about The Wire, 4th April 2008. We have been running courses which examine this claim by comparing and contrasting this fictional representation of urban America -

1 Sociology/Public Administration 342-001: Criminology Summer 1
Sociology/Public Administration 342-001: Criminology Summer 1: June 9 2014 to July 10, 2014 Online - 3 credits Instructor Office Hours Kate Gunby via email and gchat [email protected] Course Description This course begins with a quick introduction to the multidisciplinary study of criminology and how crime and criminal behavior are measured. This class explores different theories of crime and criminality, including: structural, conflict, cultural, economic, social process, psychosocial, biosocial, developmental, and broken window theories, as well as Victimology and the consequences of crime and incarceration. Throughout the class we also focus on different types of crime, including: violent crime, drug crimes, public order crime, and organized crime. This course uses the acclaimed HBO television series The Wire to explore the fundamentals of criminology. Students will develop their ability analyze, synthesize, apply, and evaluate the course material through written memos linking each reading to the content in a specific episode or film. Students will further engage with the material and each other through online forum discussions. This class is guided by student goals, which are established from the beginning and reviewed throughout the term. Readings All of the course readings are on D2L. You do not need to buy any books. Almost all of the readings are excerpts from books or articles, so please download the readings from D2L so that you only read the portions that are required for the class. Episodes and Films All of the required media for this class is available for you to stream for free on D2L. Grade Evaluation Grade Scale Introduction and Syllabus Quiz 5% A 90-100% Goals Assessments 5% B 80-89.9% Discussion Posts 30% C 70-79.9% Daily Memos 60% D 60-69.9% E 0-59.9% I do not accept late work. -

Biffy Clyro the Vertigo of Bliss Mp3, Flac, Wma
Biffy Clyro The Vertigo Of Bliss mp3, flac, wma DOWNLOAD LINKS (Clickable) Genre: Rock Album: The Vertigo Of Bliss Country: UK Released: 2003 Style: Indie Rock MP3 version RAR size: 1579 mb FLAC version RAR size: 1789 mb WMA version RAR size: 1900 mb Rating: 4.5 Votes: 568 Other Formats: MP3 MMF AC3 AA AAC AIFF ADX Tracklist 1 Bodies In Flight 5:17 2 The Ideal Height 3:41 3 With Aplomp 5:29 4 A Day Of... 2:24 5 Liberate The Illiterate 5:28 6 Diary Of Always 4:04 7 Questions & Answers 4:02 8 Eradicate The Doubt 4:26 9 When The Faction's Fractioned 3:36 10 Toys, Toys, Toys, Choke, Toys, Toys, Toys 5:28 11 All The Way Down... 6:43 12 A Man Of His Appalling Posture 3:25 13 Now The Action Is On Fire! 5:53 Other versions Category Artist Title (Format) Label Category Country Year Biffy The Vertigo Of BBQLP 233 Beggars Banquet BBQLP 233 UK 2003 Clyro Bliss (LP, Album) Beggars Banquet, Biffy The Vertigo Of Hostess BGJ-10128 BGJ-10128 Japan 2004 Clyro Bliss (CD, Album) Entertainment Unlimited none, Biffy The Vertigo Of Beggars Banquet, none, Japan 2004 KICP-986 Clyro Bliss (CD, Album) King Record Co. Ltd KICP-986 The Vertigo Of none, Biffy Beggars Banquet, none, Bliss (CD, Album, Japan 2004 DCH-15034 Clyro King Record Co. Ltd DCH-15034 Promo) The Vertigo Of Biffy Bliss (2xLP, BBQLP 2090 Beggars Banquet BBQLP 2090 UK 2013 Clyro Album, Ltd, RE, Whi) Related Music albums to The Vertigo Of Bliss by Biffy Clyro 1. -

DNA Methylation Changes in Down Syndrome Derived Neural Ipscs Uncover Co-Dysregulation of ZNF and HOX3 Families of Transcription
Laan et al. Clinical Epigenetics (2020) 12:9 https://doi.org/10.1186/s13148-019-0803-1 RESEARCH Open Access DNA methylation changes in Down syndrome derived neural iPSCs uncover co- dysregulation of ZNF and HOX3 families of transcription factors Loora Laan1†, Joakim Klar1†, Maria Sobol1, Jan Hoeber1, Mansoureh Shahsavani2, Malin Kele2, Ambrin Fatima1, Muhammad Zakaria1, Göran Annerén1, Anna Falk2, Jens Schuster1 and Niklas Dahl1* Abstract Background: Down syndrome (DS) is characterized by neurodevelopmental abnormalities caused by partial or complete trisomy of human chromosome 21 (T21). Analysis of Down syndrome brain specimens has shown global epigenetic and transcriptional changes but their interplay during early neurogenesis remains largely unknown. We differentiated induced pluripotent stem cells (iPSCs) established from two DS patients with complete T21 and matched euploid donors into two distinct neural stages corresponding to early- and mid-gestational ages. Results: Using the Illumina Infinium 450K array, we assessed the DNA methylation pattern of known CpG regions and promoters across the genome in trisomic neural iPSC derivatives, and we identified a total of 500 stably and differentially methylated CpGs that were annotated to CpG islands of 151 genes. The genes were enriched within the DNA binding category, uncovering 37 factors of importance for transcriptional regulation and chromatin structure. In particular, we observed regional epigenetic changes of the transcription factor genes ZNF69, ZNF700 and ZNF763 as well as the HOXA3, HOXB3 and HOXD3 genes. A similar clustering of differential methylation was found in the CpG islands of the HIST1 genes suggesting effects on chromatin remodeling. Conclusions: The study shows that early established differential methylation in neural iPSC derivatives with T21 are associated with a set of genes relevant for DS brain development, providing a novel framework for further studies on epigenetic changes and transcriptional dysregulation during T21 neurogenesis. -

Why Every Show Needs to Be More Like the Wire (“Not Just the Facts, Ma’Am”)
DIALOGUE WHY EVERY SHOW NEEDS TO BE MORE LIKE THE WIRE (“NOT JUST THE FACTS, MA’AM”) NEIL LANDAU University of California, Los Angeles (UCLA) The Wire (HBO, 2002-2008) upends the traditional po- ed the cop-drama universe. It was a pioneering season-long lice procedural by moving past basic plot points and “twists” procedural. Here are my top 10 reasons why Every Show in the case, diving deep into the lives of both the cops and Needs to Be More Like The Wire. the criminals they pursue. It comments on today’s America, employing characters who defy stereotype. In the words of — creator David Simon: 1. “THIS AMERICA, MAN” The grand theme here is nothing less than a nation- al existentialism: It is a police story set amid the As David Simon explains: dysfunction and indifference of an urban depart- ment—one that has failed to come to terms with In the first story arc, the episodes begin what the permanent nature of urban drug culture, one would seem to be the straightforward, albeit pro- in which thinking cops, and thinking street players, tracted, pursuit of a violent drug crew that controls must make their way independent of simple expla- a high-rise housing project. But within a brief span nations (Simon 2000: 2). of time, the officers who undertake the pursuit are forced to acknowledge truths about their de- Given the current political climate in the US and interna- partment, their role, the drug war and the city as tionally, it is timely to revisit the The Wire and how it expand- a whole. -

The Wire: a Comprehensive List of Resources
The Wire: A comprehensive list of resources Contents Introduction ............................................................................................................................ 2 W: Academic Work on The Wire........................................................................................... 3 G: General Academic Work ................................................................................................... 9 I: Wire Related Internet Sources .......................................................................................... 11 1 Introduction William Julius Wilson has argued that: "The Wire’s exploration of sociological themes is truly exceptional. Indeed I do not hesitate to say that it has done more to enhance our understandings of the challenges of urban life and urban inequality than any other media event or scholarly publication, including studies by social scientists…The Wire develops morally complex characters on each side of the law, and with its scrupulous exploration of the inner workings of various institutions, including drug-dealing gangs, the police, politicians, unions, public schools, and the print media, viewers become aware that individuals’ decisions and behaviour are often shaped by - and indeed limited by - social, political, and economic forces beyond their control". Professor William Julius Wilson, Harvard University Seminar about The Wire, 4th April 2008. We have been running courses which examine this claim by comparing and contrasting this fictional representation of urban America -

The Wire the Complete Guide
The Wire The Complete Guide PDF generated using the open source mwlib toolkit. See http://code.pediapress.com/ for more information. PDF generated at: Tue, 29 Jan 2013 02:03:03 UTC Contents Articles Overview 1 The Wire 1 David Simon 24 Writers and directors 36 Awards and nominations 38 Seasons and episodes 42 List of The Wire episodes 42 Season 1 46 Season 2 54 Season 3 61 Season 4 70 Season 5 79 Characters 86 List of The Wire characters 86 Police 95 Police of The Wire 95 Jimmy McNulty 118 Kima Greggs 124 Bunk Moreland 128 Lester Freamon 131 Herc Hauk 135 Roland Pryzbylewski 138 Ellis Carver 141 Leander Sydnor 145 Beadie Russell 147 Cedric Daniels 150 William Rawls 156 Ervin Burrell 160 Stanislaus Valchek 165 Jay Landsman 168 Law enforcement 172 Law enforcement characters of The Wire 172 Rhonda Pearlman 178 Maurice Levy 181 Street-level characters 184 Street-level characters of The Wire 184 Omar Little 190 Bubbles 196 Dennis "Cutty" Wise 199 Stringer Bell 202 Avon Barksdale 206 Marlo Stanfield 212 Proposition Joe 218 Spiros Vondas 222 The Greek 224 Chris Partlow 226 Snoop (The Wire) 230 Wee-Bey Brice 232 Bodie Broadus 235 Poot Carr 239 D'Angelo Barksdale 242 Cheese Wagstaff 245 Wallace 247 Docks 249 Characters from the docks of The Wire 249 Frank Sobotka 254 Nick Sobotka 256 Ziggy Sobotka 258 Sergei Malatov 261 Politicians 263 Politicians of The Wire 263 Tommy Carcetti 271 Clarence Royce 275 Clay Davis 279 Norman Wilson 282 School 284 School system of The Wire 284 Howard "Bunny" Colvin 290 Michael Lee 293 Duquan "Dukie" Weems 296 Namond Brice 298 Randy Wagstaff 301 Journalists 304 Journalists of The Wire 304 Augustus Haynes 309 Scott Templeton 312 Alma Gutierrez 315 Miscellany 317 And All the Pieces Matter — Five Years of Music from The Wire 317 References Article Sources and Contributors 320 Image Sources, Licenses and Contributors 324 Article Licenses License 325 1 Overview The Wire The Wire Second season intertitle Genre Crime drama Format Serial drama Created by David Simon Starring Dominic West John Doman Idris Elba Frankie Faison Larry Gilliard, Jr. -

Idioms-And-Expressions.Pdf
Idioms and Expressions by David Holmes A method for learning and remembering idioms and expressions I wrote this model as a teaching device during the time I was working in Bangkok, Thai- land, as a legal editor and language consultant, with one of the Big Four Legal and Tax companies, KPMG (during my afternoon job) after teaching at the university. When I had no legal documents to edit and no individual advising to do (which was quite frequently) I would sit at my desk, (like some old character out of a Charles Dickens’ novel) and prepare language materials to be used for helping professionals who had learned English as a second language—for even up to fifteen years in school—but who were still unable to follow a movie in English, understand the World News on TV, or converse in a colloquial style, because they’d never had a chance to hear and learn com- mon, everyday expressions such as, “It’s a done deal!” or “Drop whatever you’re doing.” Because misunderstandings of such idioms and expressions frequently caused miscom- munication between our management teams and foreign clients, I was asked to try to as- sist. I am happy to be able to share the materials that follow, such as they are, in the hope that they may be of some use and benefit to others. The simple teaching device I used was three-fold: 1. Make a note of an idiom/expression 2. Define and explain it in understandable words (including synonyms.) 3. Give at least three sample sentences to illustrate how the expression is used in context. -
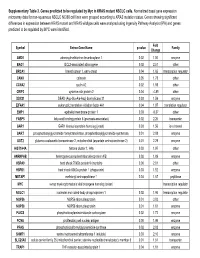
Supplementary Table 3. Genes Predicted to Be Regulated by Myc in KRAS Mutant NSCLC Cells
Supplementary Table 3. Genes predicted to be regulated by Myc in KRAS mutant NSCLC cells. Normalized basal gene expression microarray data for non-squamous NSCLC NCI60 cell lines were grouped according to KRAS mutation status. Genes showing significant differences in expression between KRAS mutant and KRAS wildtype cells were analyzed using Ingenuity Pathway Analysis (IPA) and genes predicted to be regulated by MYC were identified. Fold Symbol Entrez Gene Name p-value Family Change AMD1 adenosylmethionine decarboxylase 1 0.02 1.50 enzyme BAG1 BCL2-associated athanogene 0.02 2.57 other BRCA1 breast cancer 1, early onset 0.04 1.65 transcription regulator CANX calnexin 0.05 1.78 other CCNA2 cyclin A2 0.02 1.98 other CRIP2 cysteine-rich protein 2 0.04 -4.89 other DDX21 DEAD (Asp-Glu-Ala-Asp) box helicase 21 0.03 1.59 enzyme EIF4A1 eukaryotic translation initiation factor 4A1 0.04 1.87 translation regulator EMP1 epithelial membrane protein 1 0.03 -6.37 other FABP5 fatty acid binding protein 5 (psoriasis-associated) 0.02 2.26 transporter GAR1 GAR1 ribonucleoprotein homolog (yeast) 0.03 1.56 ion channel GART phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, 0.01 2.08 enzyme GOT2 glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) 0.01 2.29 enzyme HIST1H4A histone cluster 1, H4a 0.03 1.97 other HNRNPAB heterogeneous nuclear ribonucleoprotein A/B 0.02 1.89 enzyme HSPA9 heat shock 70kDa protein 9 (mortalin) 0.00 2.01 other HSPD1 heat shock 60kDa protein 1 (chaperonin) 0.03 1.52 enzyme -

The Justification for Protecting Reasonable Expectations
Brooklyn Law School BrooklynWorks Faculty Scholarship 2001 The uJ stification for Protecting Reasonable Expectations Bailey Kuklin Follow this and additional works at: https://brooklynworks.brooklaw.edu/faculty Part of the Law and Philosophy Commons Recommended Citation 29 Hofstra L. Rev. 863 (2001) This Article is brought to you for free and open access by BrooklynWorks. It has been accepted for inclusion in Faculty Scholarship by an authorized administrator of BrooklynWorks. THE JUSTIFICATION FOR PROTECTING REASONABLE EXPECTATIONS Bailey H. Kuklin* CONTENTS I. INTRODUCTION ......................................................................... 864 II. THE REASONS FOR PROTECTING REASONABLE EXPEcTATIONS ......................................................................... 867 A. Deontology ....................................................................... 867 B. Corrective Justice ............................................................. 869 C. DistributiveJustice ........................................................... 875 D. Intuitive Justice ................................................................. 877 E. Teleology .......................................................................... 879 F. Economics ......................................................................... 881 G. Miscellaneous................................................................... 883 Ifi. HARD CASES ............................................................................. 884 A. The Psychology of Expectations......................................