Note to Users
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Systematic Relationships and Biogeography of the Tracheophone Suboscines (Aves: Passeriformes)
MOLECULAR PHYLOGENETICS AND EVOLUTION Molecular Phylogenetics and Evolution 23 (2002) 499–512 www.academicpress.com Systematic relationships and biogeography of the tracheophone suboscines (Aves: Passeriformes) Martin Irestedt,a,b,* Jon Fjeldsaa,c Ulf S. Johansson,a,b and Per G.P. Ericsona a Department of Vertebrate Zoology and Molecular Systematics Laboratory, Swedish Museum of Natural History, P.O. Box 50007, SE-104 05 Stockholm, Sweden b Department of Zoology, University of Stockholm, SE-106 91 Stockholm, Sweden c Zoological Museum, University of Copenhagen, Copenhagen, Denmark Received 29 August 2001; received in revised form 17 January 2002 Abstract Based on their highly specialized ‘‘tracheophone’’ syrinx, the avian families Furnariidae (ovenbirds), Dendrocolaptidae (woodcreepers), Formicariidae (ground antbirds), Thamnophilidae (typical antbirds), Rhinocryptidae (tapaculos), and Conop- ophagidae (gnateaters) have long been recognized to constitute a monophyletic group of suboscine passerines. However, the monophyly of these families have been contested and their interrelationships are poorly understood, and this constrains the pos- sibilities for interpreting adaptive tendencies in this very diverse group. In this study we present a higher-level phylogeny and classification for the tracheophone birds based on phylogenetic analyses of sequence data obtained from 32 ingroup taxa. Both mitochondrial (cytochrome b) and nuclear genes (c-myc, RAG-1, and myoglobin) have been sequenced, and more than 3000 bp were subjected to parsimony and maximum-likelihood analyses. The phylogenetic signals in the mitochondrial and nuclear genes were compared and found to be very similar. The results from the analysis of the combined dataset (all genes, but with transitions at third codon positions in the cytochrome b excluded) partly corroborate previous phylogenetic hypotheses, but several novel arrangements were also suggested. -

Reference File
References added since publication of 2007 CRC Handbook of Avian Body Masses Abadie, K. B., J. Pérez Z., and M. Valverde. 2006. Primer reporte de colonias del Martín Peruano Progne murphyi. Cotinga 24:99-101. Ackerman, J. T., J. Y. Takekawa, J. D. Bluso, J. L. Yee, and C. A. Eagles-Smith. 2008. Gender identification of Caspian Terns using external morphology and discriminant function analysis. Wilson Journal of Ornithology 120:378-383. Alarcos, S., C. de la Cruz, E. Solís, J. Valencia, and M. J. García-Baquero. 2007. Sex determination of Iberian Azure-winged Magpies Cyanopica cyanus cooki by discriminant analysis of external measurements. Ringing & Migration 23:211-216. Albayrak, T., A. Besnard, and A. Erdoğan. 2011. Morphometric variation and population relationships of Krüeper’s Nuthatch (Sitta krueperi) in Turkey. Wilson Journal of Ornithology 123:734-740. Aleixo, A., C. E. B. Portes, A. Whittaker, J. D. Weckstein, L. Pedreira Gonzaga, K. J. Zimmer, C. C. Ribas, and J. M. Bates. 2013. Molecular systematics and taxonomic revision of the Curve-billed Scythebill complex (Campylorhamphus procurvoides: Dendrocolaptidae), with description of a new species from western Amazonian Brazil. Pp. 253-257, In: del Hoyo, J., A Elliott, J. Sargatal, and D.A. Christie (eds). Handbook of the birds of the world. Special volume: new species and global index. Lynx Edicions, Barcelona, Spain. Volume 1. Alfano, A. 2014. Pygmy Nightjar (Nyctopolus hirundinaeus). Neotropical Birds Online (T.S. Schulenberg, ed.). Cornell Laboratory of Ornithology, Ithaca, NY. Alvarenga, H. M. F., E. Höfling, and L. F. Silveira. 2002. Notharchus swainsoni (Gray, 1846) é uma espécie válida. -

Guia Para Observação Das Aves Do Parque Nacional De Brasília
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/234145690 Guia para observação das aves do Parque Nacional de Brasília Book · January 2011 CITATIONS READS 0 629 4 authors, including: Mieko Kanegae Fernando Lima Favaro Federal University of Rio de Janeiro Instituto Chico Mendes de Conservação da Bi… 7 PUBLICATIONS 74 CITATIONS 17 PUBLICATIONS 69 CITATIONS SEE PROFILE SEE PROFILE All content following this page was uploaded by Fernando Lima Favaro on 28 May 2014. The user has requested enhancement of the downloaded file. Brasília - 2011 GUIA PARA OBSERVAÇÃO DAS AVES DO PARQUE NACIONAL DE BRASÍLIA Aílton C. de Oliveira Mieko Ferreira Kanegae Marina Faria do Amaral Fernando de Lima Favaro Fotografia de Aves Marcelo Pontes Monteiro Nélio dos Santos Paulo André Lima Borges Brasília, 2011 GUIA PARA OBSERVAÇÃO DAS AVES DO APRESENTAÇÃO PARQUE NACIONAL DE BRASÍLIA É com grande satisfação que apresento o Guia para Observação REPÚblica FEDERATiva DO BRASIL das Aves do Parque Nacional de Brasília, o qual representa um importante instrumento auxiliar para os observadores de aves que frequentam ou que Presidente frequentarão o Parque, para fins de lazer (birdwatching), pesquisas científicas, Dilma Roussef treinamentos ou em atividades de educação ambiental. Este é mais um resultado do trabalho do Centro Nacional de Pesquisa e Vice-Presidente Conservação de Aves Silvestres - CEMAVE, unidade descentralizada do Instituto Michel Temer Chico Mendes de Conservação da Biodiversidade (ICMBio) e vinculada à Diretoria de Conservação da Biodiversidade. O Centro tem como missão Ministério do Meio Ambiente - MMA subsidiar a conservação das aves brasileiras e dos ambientes dos quais elas Izabella Mônica Vieira Teixeira dependem. -

ABSTRACT BOOK Listed Alphabetically by Last Name Of
ABSTRACT BOOK Listed alphabetically by last name of presenting author AOS 2019 Meeting 24-28 June 2019 ORAL PRESENTATIONS Variability in the Use of Acoustic Space Between propensity, renesting intervals, and renest reproductive Two Tropical Forest Bird Communities success of Piping Plovers (Charadrius melodus) by fol- lowing 1,922 nests and 1,785 unique breeding adults Patrick J Hart, Kristina L Paxton, Grace Tredinnick from 2014 2016 in North and South Dakota, USA. The apparent renesting rate was 20%. Renesting propen- When acoustic signals sent from individuals overlap sity declined if reproductive attempts failed during the in frequency or time, acoustic interference and signal brood-rearing stage, nests were depredated, reproduc- masking occurs, which may reduce the receiver’s abil- tive failure occurred later in the breeding season, or ity to discriminate information from the signal. Under individuals had previously renested that year. Addi- the acoustic niche hypothesis (ANH), acoustic space is tionally, plovers were less likely to renest on reservoirs a resource that organisms may compete for, and sig- compared to other habitats. Renesting intervals de- naling behavior has evolved to minimize overlap with clined when individuals had not already renested, were heterospecific calling individuals. Because tropical after second-year adults without prior breeding experi- wet forests have such high bird species diversity and ence, and moved short distances between nest attempts. abundance, and thus high potential for competition for Renesting intervals also decreased if the attempt failed acoustic niche space, they are good places to examine later in the season. Lastly, overall reproductive success the way acoustic space is partitioned. -

An Update of Wallacels Zoogeographic Regions of the World
REPORTS To examine the temporal profile of ChC produc- specification of a distinct, and probably the last, 3. G. A. Ascoli et al., Nat. Rev. Neurosci. 9, 557 (2008). tion and their correlation to laminar deployment, cohort in this lineage—the ChCs. 4. J. Szentágothai, M. A. Arbib, Neurosci. Res. Program Bull. 12, 305 (1974). we injected a single pulse of BrdU into pregnant A recent study demonstrated that progeni- CreER 5. P. Somogyi, Brain Res. 136, 345 (1977). Nkx2.1 ;Ai9 females at successive days be- tors below the ventral wall of the lateral ventricle 6. L. Sussel, O. Marin, S. Kimura, J. L. Rubenstein, tween E15 and P1 to label mitotic progenitors, (i.e., VGZ) of human infants give rise to a medial Development 126, 3359 (1999). each paired with a pulse of tamoxifen at E17 to migratory stream destined to the ventral mPFC 7. S. J. Butt et al., Neuron 59, 722 (2008). + 18 8. H. Taniguchi et al., Neuron 71, 995 (2011). label NKX2.1 cells (Fig. 3A). We first quanti- ( ). Despite species differences in the develop- 9. L. Madisen et al., Nat. Neurosci. 13, 133 (2010). fied the fraction of L2 ChCs (identified by mor- mental timing of corticogenesis, this study and 10. J. Szabadics et al., Science 311, 233 (2006). + phology) in mPFC that were also BrdU+. Although our findings raise the possibility that the NKX2.1 11. A. Woodruff, Q. Xu, S. A. Anderson, R. Yuste, Front. there was ChC production by E15, consistent progenitors in VGZ and their extended neurogenesis Neural Circuits 3, 15 (2009). -

A Study of the Rufous-Fronted Thornbird and Associated Birds
A STUDY OF THE RUFOUS-FRONTED THORNBIRD AND ASSOCIATED BIRDS ALEXANDER F. SKUTCH ANGING from isolated trees in northern Venezuela are compact masses of H interlaced twigs so large and conspicuous that they can hardly escape the attention even of the traveler speeding along the excellent highways of that country. From a Venezuelan companion he may learn that the oblong masses of sticks are nests of the guaiti; but without patient watching he is not likely to see the retiring, wren-like builder of these incongruously large structures. The guaiti bears the English book-name of Rufous-fronted Thorn- bird (Phacellodomus rufifrons) and is a member of a large Neotropical family, the Furnariidae or ovenbirds, second to no other avian family in the world in the diversity of its nidification. The brief accounts that I had read of this remarkable bird, suggesting unusual social habits, whetted my desire to learn more about it. A grant from the Frank M. Chapman Memorial Fund of the American Museum of Natural History provided financial support for an extended visit to Venezuela. Cor- respondence with Paul Schwartz gave hope that we could find a suitable place to live while studying thornbirds. He met us at Maiquetia Airport soon after midnight on 12 March 1966 and, after showing us over the Parque National “Henri Pittier” (Ranch0 Grande), took us to “La Araguata.” This large cattle farm, belonging to the bird artist Walter Arp and his wife Elena, is situated, at an elevation around 1,400 feet, in a basin among low mountains near Pirapira, about 20 miles south of Valencia in the state of Carabobo. -

IGUAZU FALLS Extension 1-15 December 2016
Tropical Birding Trip Report NW Argentina & Iguazu Falls: December 2016 A Tropical Birding SET DEPARTURE tour NW ARGENTINA: High Andes, Yungas and Monte Desert and IGUAZU FALLS Extension 1-15 December 2016 TOUR LEADER: ANDRES VASQUEZ (All Photos by Andres Vasquez) A combination of breathtaking landscapes and stunning birds are what define this tour. Clockwise from bottom left: Cerro de los 7 Colores in the Humahuaca Valley, a World Heritage Site; Wedge-tailed Hillstar at Yavi; Ochre-collared Piculet on the Iguazu Falls Extension; and one of the innumerable angles of one of the World’s-must-visit destinations, Iguazu Falls. www.tropicalbirding.com +1-409-515-9110 [email protected] p.1 Tropical Birding Trip Report NW Argentina & Iguazu Falls: December 2016 Introduction: This is the only tour that I guide where I feel that the scenery is as impressive (or even surpasses) the birds themselves. This is not to say that the birds are dull on this tour, far from it. Some of the avian highlights included wonderfully jeweled hummingbirds like Wedge-tailed Hillstar and Red-tailed Comet; getting EXCELLENT views of 4 Tinamou species of, (a rare thing on all South American tours except this one); nearly 20 species of ducks, geese and swans, with highlights being repeated views of Torrent Ducks, the rare and oddly, parasitic Black-headed Duck, the beautiful Rosy-billed Pochard, and the mountain-dwelling Andean Goose. And we should not forget other popular bird features like 3 species of Flamingos on one lake, 11 species of Woodpeckers, including the hulking Cream-backed, colorful Yellow-fronted and minuscule Ochre-collared Piculet on the extension to Iguazu Falls. -

Supplemental Wing Shape and Dispersal Analysis
Data Supplement High dispersal ability inhibits speciation in a continental radiation of passerine birds Santiago Claramunt, Elizabeth P. Derryberry, J. V. Remsen, Jr. & Robb T. Brumfield Museum of Natural Science and Department of Biological Sciences, Louisiana State University, Baton Rouge, LA 70803, USA HAND-WING INDEX AND FLIGHT PERFORMANCE IN NEOTROPICAL FOREST BIRDS We investigated the relationship between wing shape and flight distances determined during 'dispersal challenge' experiments conducted in Gatun Lake in the Panama Canal (Moore et al. 2008). During the experiments, birds were released from a boat at incremental distances from shore and the distance flown or the success or failure in reaching the coast was recorded. To investigated the relationship between the hand-wing index and flight distance in Neotropical birds we used data on mean distance flown from table 3 in ref. We estimated hand-wing indices for the 10 species reported in those experiments (Table S1) . Wing measurements were taken by SC for four males of each species at LSUMNS. The relationship between the hand-wing index and distance flown was evaluated statistically using phylogenetic generalized least-squares (PGLS, Freckleton et al. 2002). We generated a phylogeny for the species involved in the experiment or an appropriate surrogate using DNA sequences of the slow-evolving RAG 1 gene from GenBank (Table S2). A maximum likelihood ultrametric tree was generated in PAUP* (Swofford 2003) using a GTR+! model of nucleotide substitution rates, empirical nucleotide frequencies, and enforcing a molecular clock. We found that the hand-wing index was strongly related to mean distance flown (R2 = 0.68, F = 20, d.f. -

Evolution of the Ovenbird-Woodcreeper Assemblage (Aves: Furnariidae) Б/ Major Shifts in Nest Architecture and Adaptive Radiatio
JOURNAL OF AVIAN BIOLOGY 37: 260Á/272, 2006 Evolution of the ovenbird-woodcreeper assemblage (Aves: Furnariidae) / major shifts in nest architecture and adaptive radiation Á Martin Irestedt, Jon Fjeldsa˚ and Per G. P. Ericson Irestedt, M., Fjeldsa˚, J. and Ericson, P. G. P. 2006. Evolution of the ovenbird- woodcreeper assemblage (Aves: Furnariidae) Á/ major shifts in nest architecture and adaptive radiation. Á/ J. Avian Biol. 37: 260Á/272 The Neotropical ovenbirds (Furnariidae) form an extraordinary morphologically and ecologically diverse passerine radiation, which includes many examples of species that are superficially similar to other passerine birds as a resulting from their adaptations to similar lifestyles. The ovenbirds further exhibits a truly remarkable variation in nest types, arguably approaching that found in the entire passerine clade. Herein we present a genus-level phylogeny of ovenbirds based on both mitochondrial and nuclear DNA including a more complete taxon sampling than in previous molecular studies of the group. The phylogenetic results are in good agreement with earlier molecular studies of ovenbirds, and supports the suggestion that Geositta and Sclerurus form the sister clade to both core-ovenbirds and woodcreepers. Within the core-ovenbirds several relationships that are incongruent with traditional classifications are suggested. Among other things, the philydorine ovenbirds are found to be non-monophyletic. The mapping of principal nesting strategies onto the molecular phylogeny suggests cavity nesting to be plesiomorphic within the ovenbirdÁ/woodcreeper radiation. It is also suggested that the shift from cavity nesting to building vegetative nests is likely to have happened at least three times during the evolution of the group. -

Phylogenetic Analysis of the Nest Architecture of Neotropical Ovenbirds (Furnariidae)
The Auk 116(4):891-911, 1999 PHYLOGENETIC ANALYSIS OF THE NEST ARCHITECTURE OF NEOTROPICAL OVENBIRDS (FURNARIIDAE) KRZYSZTOF ZYSKOWSKI • AND RICHARD O. PRUM NaturalHistory Museum and Department of Ecologyand Evolutionary Biology, University of Kansas,Lawrence, Kansas66045, USA ABSTRACT.--Wereviewed the tremendousarchitectural diversity of ovenbird(Furnari- idae) nestsbased on literature,museum collections, and new field observations.With few exceptions,furnariids exhibited low intraspecificvariation for the nestcharacters hypothe- sized,with the majorityof variationbeing hierarchicallydistributed among taxa. We hy- pothesizednest homologies for 168species in 41 genera(ca. 70% of all speciesand genera) and codedthem as 24 derivedcharacters. Forty-eight most-parsimonious trees (41 steps,CI = 0.98, RC = 0.97) resultedfrom a parsimonyanalysis of the equallyweighted characters using PAUP,with the Dendrocolaptidaeand Formicarioideaas successiveoutgroups. The strict-consensustopology based on thesetrees contained 15 cladesrepresenting both tra- ditionaltaxa and novelphylogenetic groupings. Comparisons with the outgroupsdemon- stratethat cavitynesting is plesiomorphicto the furnariids.In the two lineageswhere the primitivecavity nest has been lost, novel nest structures have evolved to enclosethe nest contents:the clayoven of Furnariusand the domedvegetative nest of the synallaxineclade. Althoughour phylogenetichypothesis should be consideredas a heuristicprediction to be testedsubsequently by additionalcharacter evidence, this first cladisticanalysis -

On the Origin and Evolution of Nest Building by Passerine Birds’
T H E C 0 N D 0 R r : : ,‘ “; i‘ . .. \ :i A JOURNAL OF AVIAN BIOLOGY ,I : Volume 99 Number 2 ’ I _ pg$$ij ,- The Condor 99~253-270 D The Cooper Ornithological Society 1997 ON THE ORIGIN AND EVOLUTION OF NEST BUILDING BY PASSERINE BIRDS’ NICHOLAS E. COLLIAS Departmentof Biology, Universityof California, Los Angeles, CA 90024-1606 Abstract. The object of this review is to relate nest-buildingbehavior to the origin and early evolution of passerinebirds (Order Passeriformes).I present evidence for the hypoth- esis that the combinationof small body size and the ability to place a constructednest where the bird chooses,helped make possiblea vast amountof adaptiveradiation. A great diversity of potential habitats especially accessibleto small birds was created in the late Tertiary by global climatic changes and by the continuing great evolutionary expansion of flowering plants and insects.Cavity or hole nests(in ground or tree), open-cupnests (outside of holes), and domed nests (with a constructedroof) were all present very early in evolution of the Passeriformes,as indicated by the presenceof all three of these basic nest types among the most primitive families of living passerinebirds. Secondary specializationsof these basic nest types are illustratedin the largest and most successfulfamilies of suboscinebirds. Nest site and nest form and structureoften help characterizethe genus, as is exemplified in the suboscinesby the ovenbirds(Furnariidae), a large family that builds among the most diverse nests of any family of birds. The domed nest is much more common among passerinesthan in non-passerines,and it is especially frequent among the very smallestpasserine birds the world over. -
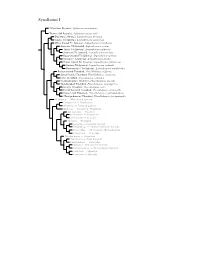
Synallaxini Species Tree
Synallaxini I ?Masafuera Rayadito, Aphrastura masafuerae Thorn-tailed Rayadito, Aphrastura spinicauda Des Murs’s Wiretail, Leptasthenura desmurii Tawny Tit-Spinetail, Leptasthenura yanacensis White-browed Tit-Spinetail, Leptasthenura xenothorax Araucaria Tit-Spinetail, Leptasthenura setaria Tufted Tit-Spinetail, Leptasthenura platensis Striolated Tit-Spinetail, Leptasthenura striolata Rusty-crowned Tit-Spinetail, Leptasthenura pileata Streaked Tit-Spinetail, Leptasthenura striata Brown-capped Tit-Spinetail, Leptasthenura fuliginiceps Andean Tit-Spinetail, Leptasthenura andicola Plain-mantled Tit-Spinetail, Leptasthenura aegithaloides Rufous-fronted Thornbird, Phacellodomus rufifrons Streak-fronted Thornbird, Phacellodomus striaticeps Little Thornbird, Phacellodomus sibilatrix Chestnut-backed Thornbird, Phacellodomus dorsalis Spot-breasted Thornbird, Phacellodomus maculipectus Greater Thornbird, Phacellodomus ruber Freckle-breasted Thornbird, Phacellodomus striaticollis Orange-eyed Thornbird, Phacellodomus erythrophthalmus ?Orange-breasted Thornbird, Phacellodomus ferrugineigula Hellmayrea — White-tailed Spinetail Coryphistera — Brushrunner Anumbius — Firewood-gatherer Asthenes — Canasteros, Thistletails Acrobatornis — Graveteiro Metopothrix — Plushcrown Xenerpestes — Graytails Siptornis — Prickletail Roraimia — Roraiman Barbtail Thripophaga — Speckled Spinetail, Softtails Limnoctites — ST Spinetail, SB Reedhaunter Cranioleuca — Spinetails Pseudasthenes — Canasteros Spartonoica — Wren-Spinetail Pseudoseisura — Cacholotes Mazaria – White-bellied