Umeå University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Prediction of Meiosis-Essential Genes Based Upon the Dynamic Proteomes Responsive to Spermatogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.05.936435; this version posted February 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Prediction of meiosis-essential genes based upon the dynamic proteomes responsive to spermatogenesis Kailun Fang1,2,3,8, Qidan Li3,4,5,8, Yu Wei2,3,8, Jiaqi Shen6,8, Wenhui Guo3,4,5,7,8, Changyang Zhou2,3,8, Ruoxi Wu1, Wenqin Ying2, Lu Yu1,2, Jin Zi5, Yuxing Zhang3,4,5, Hui Yang2,3,9*, Siqi Liu3,4,5,9*, Charlie Degui Chen1,3,9* 1. State Key Laboratory of Molecular Biology, Shanghai Key Laboratory of Molecular Andrology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai 200031, China. 2. State Key Laboratory of Neuroscience, Key Laboratory of Primate Neurobiology, CAS Center for Excellence in Brain Science and Intelligence Technology, Shanghai Research Center for Brain Science and Brain-Inspired Intelligence, Institute of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China. 3. University of the Chinese Academy of Sciences, Beijing 100049, China. 4. CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China. 5. BGI Genomics, BGI-Shenzhen, Shenzhen 518083, China. 6. United World College Changshu China, Jiangsu 215500, China. 7. Malvern College Qingdao, Shandong, 266109, China 8. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Construction and Initial Characterization of the Densin
Chapter 2: Design of Targeting Construct for Densin Deletion, Confirmation of Densin Knockout, and Initial Characterization of the Knockout Phenotype Introduction Derangements in synaptic transmission and plasticity are part of the pathology of numerous neurological and mental health diseases including epilepsy, schizophrenia, depression, and Alzheimer’s disease. In excitatory synapses of the CNS, the postsynaptic reception, integration, and transduction of signals is mediated by the supermolecular complex of the postsynaptic density. Understanding the role that particular PSD proteins play in normal and pathological states will greatly enhance our knowledge of the underlying molecular mechanisms which contribute to overall mental health and well being. A major step in the study of a protein’s function in any biological system is the generation of a mutant phenotype that completely lacks expression of the protein. Numerous core proteins of the PSD have been studied in this manner, including PSD-95 [1], CaMKII [2, 3], the GluR2 subunit of the AMPA receptor [4], SynGAP [5], - catenin [6], and Shank [7]. Knockouts have also been generated for all the subunits of the NMDA receptor, including the NR1 subunit [8, 9], NR2A subunit [10], NR2B subunit [11], NR2C subunit [12] and NR2D subunit [13]. Finally, transgenic animals have been generated with deletions in the cytoplasmic tails of the NR2A, NR2B, and 29 NR2C subunits of the NMDA receptor [14]. These mutant and transgenic animals have provided an immensely detailed understanding of their roles in synaptic transmission and plasticity. However, a more holistic understanding of how these core PSD proteins are functionally and structurally integrated into the supramolecular complex of the PSD still remains elusive. -

Gene Interaction Analysis for Next-Generation Sequencing
European Journal of Human Genetics (2016) 24, 421–428 & 2016 Macmillan Publishers Limited All rights reserved 1018-4813/16 www.nature.com/ejhg ARTICLE Genome-wide gene–gene interaction analysis for next-generation sequencing Jinying Zhao1, Yun Zhu1 and Momiao Xiong*,2 The critical barrier in interaction analysis for next-generation sequencing (NGS) data is that the traditional pairwise interaction analysis that is suitable for common variants is difficult to apply to rare variants because of their prohibitive computational time, large number of tests and low power. The great challenges for successful detection of interactions with NGS data are (1) the demands in the paradigm of changes in interaction analysis; (2) severe multiple testing; and (3) heavy computations. To meet these challenges, we shift the paradigm of interaction analysis between two SNPs to interaction analysis between two genomic regions. In other words, we take a gene as a unit of analysis and use functional data analysis techniques as dimensional reduction tools to develop a novel statistic to collectively test interaction between all possible pairs of SNPs within two genome regions. By intensive simulations, we demonstrate that the functional logistic regression for interaction analysis has the correct type 1 error rates and higher power to detect interaction than the currently used methods. The proposed method was applied to a coronary artery disease dataset from the Wellcome Trust Case Control Consortium (WTCCC) study and the Framingham Heart Study (FHS) dataset, and the early-onset myocardial infarction (EOMI) exome sequence datasets with European origin from the NHLBI’s Exome Sequencing Project. We discovered that 6 of 27 pairs of significantly interacted genes in the FHS were replicated in the independent WTCCC study and 24 pairs of significantly interacted genes after applying Bonferroni correction in the EOMI study. -

Functionally Enigmatic Genes: a Case Study of the Brain Ignorome
Functionally Enigmatic Genes: A Case Study of the Brain Ignorome Ashutosh K. Pandey1*,LuLu1, Xusheng Wang1,2, Ramin Homayouni3, Robert W. Williams1 1 UT Center for Integrative and Translational Genomics and Department of Anatomy and Neurobiology, University of Tennessee Health Science Center, Memphis, Tennessee, United States of America, 2 St. Jude Children’s Research Hospital, Memphis, Tennessee, United States of America, 3 Department of Biological Sciences, Bioinformatics Program, University of Memphis, Memphis, Tennessee, United States of America Abstract What proportion of genes with intense and selective expression in specific tissues, cells, or systems are still almost completely uncharacterized with respect to biological function? In what ways do these functionally enigmatic genes differ from well-studied genes? To address these two questions, we devised a computational approach that defines so-called ignoromes. As proof of principle, we extracted and analyzed a large subset of genes with intense and selective expression in brain. We find that publications associated with this set are highly skewed—the top 5% of genes absorb 70% of the relevant literature. In contrast, approximately 20% of genes have essentially no neuroscience literature. Analysis of the ignorome over the past decade demonstrates that it is stubbornly persistent, and the rapid expansion of the neuroscience literature has not had the expected effect on numbers of these genes. Surprisingly, ignorome genes do not differ from well-studied genes in terms of connectivity in coexpression networks. Nor do they differ with respect to numbers of orthologs, paralogs, or protein domains. The major distinguishing characteristic between these sets of genes is date of discovery, early discovery being associated with greater research momentum—a genomic bandwagon effect. -
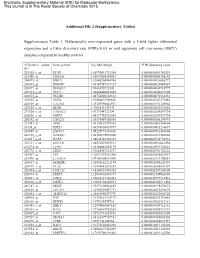
Differentially Over-Expressed Genes with a 2-Fold Higher Differe
Electronic Supplementary Material (ESI) for Molecular BioSystems. This journal is © The Royal Society of Chemistry 2015 Additional File 2 (Supplementary Tables) Supplementary Table 1: Differentially over-expressed genes with a 2-fold higher differential expression and a False discovery rate (FDR)<0.05 in oral squamous cell carcinoma (OSCC) samples compared to healthy controls. Affymetrix probe Gene symbol Log fold change FDR adjusted-p value ID 209365_s_at ECM1 1.65970411718384 0.0000000001342029 211980_at COL4A1 1.63093004254041 0.0000000003388142 204475_at MMP1 1.21640208064746 0.0000000016668772 206605_at ENDOU 1.49187987837927 0.0000000020488567 218677_at S100A14 1.5666356921542 0.0000000030718997 201325_s_at EMP1 1.42488420822805 0.0000000042431990 203675_at NUCB2 1.48754002522513 0.0000000076914233 218935_at EHD3 1.51884627509609 0.0000000147373416 220149_at C2orf54 1.35307996462937 0.0000000151328942 207655_s_at BLNK 1.4981534243447 0.0000000230316858 217508_s_at C18orf25 1.4737344722194 0.0000000259591734 203936_s_at MMP9 1.54117752982424 0.0000000259591734 205242_at CXCL13 1.36176459188585 0.0000000265290993 212543_at AIM1 1.43105615599363 0.0000000400764664 38158_at ESPL1 1.43594803693999 0.0000000415216687 203585_at ZNF185 1.54225773236818 0.0000000521483660 220330_s_at SAMSN1 1.48385675070206 0.0000000531560858 212657_s_at IL1RN 1.38628748328333 0.0000000754734933 202311_s_at COL1A1 1.44526073425523 0.0000000810641434 212274_at LPIN1 1.28166083845179 0.0000000951352623 209772_s_at CD24 1.39648587121677 0.0000000951352623 203407_at -

Enrichment of Mutations in Chromatin Regulators in People with Rett Syndrome Lacking Mutations in MECP2
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Genet Med Manuscript Author . Author manuscript; Manuscript Author available in PMC 2016 November 14. Enrichment of mutations in chromatin regulators in people with Rett Syndrome lacking mutations in MECP2 Samin A. Sajan, PhD1,2,#, Shalini N. Jhangiani, MS3, Donna M. Muzny, MS3, Richard A. Gibbs, PhD3,4, James R. Lupski, MD, PhD3,4,5, Daniel G. Glaze, MD1, Walter E. Kaufmann, MD6, Steven A. Skinner, MD7, Fran Anese7, Michael J. Friez, PhD7, Lane Jane, RN8, Alan K. Percy, MD8, and Jeffrey L. Neul, MD, PhD1,2,4,# 1Section of Child Neurology and Developmental Neuroscience, Department of Pediatrics, Baylor College of Medicine, Houston, Texas, USA 2Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital, Houston, Texas, USA 3Human Genome Sequencing Center, Baylor College of Medicine, Houston, Texas, USA 4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas, USA 5Department of Pediatrics, Baylor College of Medicine and Texas Children's Hospital, Houston, Texas, USA 6Department of Neurology, Boston Children's Hospital, Boston, Massachusetts, USA 7Greenwood Genetic Center, Greenwood, South Carolina, USA 8Department of Pediatrics, University of Alabama at Birmingham, Birmingham, Alabama, USA Abstract Purpose—Rett Syndrome (RTT) is a neurodevelopmental disorder caused primarily by de novo mutations (DNMs) in MECP2 and sometimes in CDKL5 and FOXG1. However, some RTT cases lack mutations in these genes. Methods—Twenty-two RTT cases without apparent MECP2, CDKL5, and FOXG1 mutations were subjected to both whole exome sequencing and single nucleotide polymorphism array-based copy number variant (CNV) analyses. Results—Three cases had MECP2 mutations initially missed by clinical testing. -

A Second Generation Human Haplotype Map of Over 3.1 Million Snps the International Hapmap Consortium1
doi: 10.1038/nature06258 SUPPLEMENTARY INFORMATION A second generation human haplotype map of over 3.1 million SNPs The International HapMap Consortium1 Supplementary material S1 The density of common SNPs in the Phase II HapMap and the assembled human genome S2 Analysis of data quality S2.1 Analysis of amplicon structure to genotyping error S2.2 Analysis of genotype discordance from overlap with Seattle SNPs S2.3 Analysis of genotype discordance from fosmid end sequences S2.4 Analysis of monomorphism/polymorphism discrepancies S2.5 Interchromosomal LD S3. Analysis of population stratification S4. Analysis of relatedness S5. Segmental analysis of relatedness S6. Analysis of homozygosity S7. Perlegen genotyping protocols Supplementary tables Legends to supplementary figures 1 See end of manuscript for Consortium details www.nature.com/nature 1 doi: 10.1038/nature06258 SUPPLEMENTARY INFORMATION Supplementary text 1. The density of common SNPs in the Phase II HapMap and the assembled human genome. To estimate the fraction of all common variants on the autosomes that have been successfully genotyped in the consensus Phase II HapMap we note that in YRI (release 21) there are 2,334,980 SNPs with MAF≥0.05. Across the autosomes, the completed reference sequence assembled in contigs is 2.68 billion bp. Assuming that the allele frequency distribution in the YRI is well approximated by that of a simple coalescent model and using an estimate of the population mutation rate of θ = 1.2 per kb for African populations1,2 the expected number of variants with MAF≥0.05 in a sample of 120 chromosomes is 114 = θ E(S MAF≥5% ) L ∑ /1 i i=6 where L is the total length of the sequence3. -

The Role of Lamin Associated Domains in Global Chromatin Organization and Nuclear Architecture
THE ROLE OF LAMIN ASSOCIATED DOMAINS IN GLOBAL CHROMATIN ORGANIZATION AND NUCLEAR ARCHITECTURE By Teresa Romeo Luperchio A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland March 2016 © 2016 Teresa Romeo Luperchio All Rights Reserved ABSTRACT Nuclear structure and scaffolding have been implicated in expression and regulation of the genome (Elcock and Bridger 2010; Fedorova and Zink 2008; Ferrai et al. 2010; Li and Reinberg 2011; Austin and Bellini 2010). Discrete domains of chromatin exist within the nuclear volume, and are suggested to be organized by patterns of gene activity (Zhao, Bodnar, and Spector 2009). The nuclear periphery, which consists of the inner nuclear membrane and associated proteins, forms a sub- nuclear compartment that is mostly associated with transcriptionally repressed chromatin and low gene expression (Guelen et al. 2008). Previous studies from our lab and others have shown that repositioning genes to the nuclear periphery is sufficient to induce transcriptional repression (K L Reddy et al. 2008; Finlan et al. 2008). In addition, a number of studies have provided evidence that many tissue types, including muscle, brain and blood, use the nuclear periphery as a compartment during development to regulate expression of lineage specific genes (Meister et al. 2010; Szczerbal, Foster, and Bridger 2009; Yao et al. 2011; Kosak et al. 2002; Peric-Hupkes et al. 2010). These large regions of chromatin that come in molecular contact with the nuclear periphery are called Lamin Associated Domains (LADs). The studies described in this dissertation have furthered our understanding of maintenance and establishment of LADs as well as the relationship of LADs with the epigenome and other factors that influence three-dimensional chromatin structure. -

Supplemental Solier
Supplementary Figure 1. Importance of Exon numbers for transcript downregulation by CPT Numbers of down-regulated genes for four groups of comparable size genes, differing only by the number of exons. Supplementary Figure 2. CPT up-regulates the p53 signaling pathway genes A, List of the GO categories for the up-regulated genes in CPT-treated HCT116 cells (p<0.05). In bold: GO category also present for the genes that are up-regulated in CPT- treated MCF7 cells. B, List of the up-regulated genes in both CPT-treated HCT116 cells and CPT-treated MCF7 cells (CPT 4 h). C, RT-PCR showing the effect of CPT on JUN and H2AFJ transcripts. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. Supplementary Figure 3. Down-regulation of RNA degradation-related genes after CPT treatment A, “RNA degradation” pathway from KEGG. The genes with “red stars” were down- regulated genes after CPT treatment. B, Affy Exon array data for the “CNOT” genes. The log2 difference for the “CNOT” genes expression depending on CPT treatment was normalized to the untreated controls. C, RT-PCR showing the effect of CPT on “CNOT” genes down-regulation. HCT116 cells were treated with CPT (10 µM, 20 h) and CNOT6L, CNOT2, CNOT4 and CNOT6 mRNA were analysed by RT-PCR. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. D, CNOT6L down-regulation after CPT treatment. CNOT6L transcript was analysed by Q- PCR. Supplementary Figure 4. Down-regulation of ubiquitin-related genes after CPT treatment A, “Ubiquitin-mediated proteolysis” pathway from KEGG.