Structural and Mechanistic Investigations on Salmonella
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
Supplementary Materials Figure S1. Differentially abundant spots between the mid-log phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 3. Figure S2. Differentially abundant spots between the stationary phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 4. S2 Table S1. Summary of the non-polysaccharide degrading proteins identified in the B. proteoclasticus cytosol by 2DE/MALDI-TOF. Protein Locus Location Score pI kDa Pep. Cov. Amino Acid Biosynthesis Acetylornithine aminotransferase, ArgD Bpr_I1809 C 1.7 × 10−4 5.1 43.9 11 34% Aspartate/tyrosine/aromatic aminotransferase Bpr_I2631 C 3.0 × 10−14 4.7 43.8 15 46% Aspartate-semialdehyde dehydrogenase, Asd Bpr_I1664 C 7.6 × 10−18 5.5 40.1 17 50% Branched-chain amino acid aminotransferase, IlvE Bpr_I1650 C 2.4 × 10−12 5.2 39.2 13 32% Cysteine synthase, CysK Bpr_I1089 C 1.9 × 10−13 5.0 32.3 18 72% Diaminopimelate dehydrogenase Bpr_I0298 C 9.6 × 10−16 5.6 35.8 16 49% Dihydrodipicolinate reductase, DapB Bpr_I2453 C 2.7 × 10−6 4.9 27.0 9 46% Glu/Leu/Phe/Val dehydrogenase Bpr_I2129 C 1.2 × 10−30 5.4 48.6 31 64% Imidazole glycerol phosphate synthase Bpr_I1240 C 8.0 × 10−3 4.7 22.5 8 44% glutamine amidotransferase subunit Ketol-acid reductoisomerase, IlvC Bpr_I1657 C 3.8 × 10−16 -

Detoxification of Lignocellulose-Derived Microbial Inhibitory Compounds by Clostridium Beijerinckii NCIMB 8052 During Acetone-Butanol-Ethanol Fermentation
Detoxification of Lignocellulose-derived Microbial Inhibitory Compounds by Clostridium beijerinckii NCIMB 8052 during Acetone-Butanol-Ethanol Fermentation DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Yan Zhang Graduate Program in Animal Sciences The Ohio State University 2013 Dissertation Committee: Thaddeus C. Ezeji, Advisor Steven C. Loerch Sandra G. Velleman Zhongtang Yu Venkat Gopalan Copyrighted by Yan Zhang 2013 Abstract Pretreatment and hydrolysis of lignocellulosic biomass to fermentable sugars generate a complex mixture of microbial inhibitors such as furan aldehydes (e.g., furfural), which at sublethal concentration in the fermentation medium can be tolerated or detoxified by acetone butanol ethanol (ABE)-producing Clostridium beijerinckii NCIMB 8052. The response of C. beijerinckii to furfural at the molecular level, however, has not been directly studied. Therefore, this study was to elucidate mechanism employed by C. beijerinckii to detoxify lignocellulose-derived microbial inhibitors and use this information to develop inhibitor-tolerant C. beijerinckii. Towards the long-term goal of developing inhibitor-tolerant Clostridium strains, the first objective was to evaluate ABE fermentation by C. beijerinckii using different proportions of Miscanthus giganteus hydrolysates as carbon source. Compared to the growth of C. beijerinckii in control medium, C. beijerinckii experienced different degrees of inhibition. The degree of inhibition was dose-dependent, and C. beijerinckii did not grow in P2 medium with greater than 25% (v/v) Miscanthus giganteus hydrolysates. To improve tolerance of C. beijerinckii to inhibitors, supplementation of P2 medium with undiluted (100%) Miscanthus giganteus hydrolysates with 4 g/L CaCO3 resulted in successful growth of and ABE production by C. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Production of Butyric Acid and Hydrogen by Metabolically Engineered Mutants of Clostridium Tyrobutyricum
PRODUCTION OF BUTYRIC ACID AND HYDROGEN BY METABOLICALLY ENGINEERED MUTANTS OF CLOSTRIDIUM TYROBUTYRICUM DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Xiaoguang Liu, M.S. ***** The Ohio State University 2005 Dissertation committee: Approved by Professor Shang-Tian Yang, Adviser Professor Barbara E Wyslouzil Adviser Professor Hua Wang Department of Chemical Engineering ABSTRACT Butyric acid has many applications in chemical, food and pharmaceutical industries. The production of butyric acid by fermentation has become an increasingly attractive alternative to current petroleum-based chemical synthesis. Clostridium tyrobutyricum is an anaerobic bacterium producing butyric acid, acetic acid, hydrogen and carbon dioxide as its main products. Hydrogen, as an energy byproduct, can add value to the fermentation process. The goal of this project was to develop novel bioprocess to produce butyric acid and hydrogen economically by Clostridial mutants. Conventional fermentation technologies for butyric acid and hydrogen production are limited by low reactor productivity, product concentration and yield. In this project, novel engineered mutants of C. tyrobutyricum were created by gene manipulation and cell adaptation. Fermentation process was also optimized using immobilizing cells in the fibrous-bed bioreactor (FBB) to enhance butyric acid and hydrogen production. First, metabolic engineered mutants with knocked-out acetate formation pathway were created and characterized. Gene inactivation technology was used to delete the genes of phosphotransacetylase (PTA) and acetate kinase (AK), two key enzymes in the acetate-producing pathway of C. tyrobutyricum, through homologous recombination. The metabolic engineered mutants were characterized by Southern hybridization, enzyme assay, protein expression and metabolites production. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

The Metabolic Building Blocks of a Minimal Cell Supplementary
The metabolic building blocks of a minimal cell Mariana Reyes-Prieto, Rosario Gil, Mercè Llabrés, Pere Palmer and Andrés Moya Supplementary material. Table S1. List of enzymes and reactions modified from Gabaldon et. al. (2007). n.i.: non identified. E.C. Name Reaction Gil et. al. 2004 Glass et. al. 2006 number 2.7.1.69 phosphotransferase system glc + pep → g6p + pyr PTS MG041, 069, 429 5.3.1.9 glucose-6-phosphate isomerase g6p ↔ f6p PGI MG111 2.7.1.11 6-phosphofructokinase f6p + atp → fbp + adp PFK MG215 4.1.2.13 fructose-1,6-bisphosphate aldolase fbp ↔ gdp + dhp FBA MG023 5.3.1.1 triose-phosphate isomerase gdp ↔ dhp TPI MG431 glyceraldehyde-3-phosphate gdp + nad + p ↔ bpg + 1.2.1.12 GAP MG301 dehydrogenase nadh 2.7.2.3 phosphoglycerate kinase bpg + adp ↔ 3pg + atp PGK MG300 5.4.2.1 phosphoglycerate mutase 3pg ↔ 2pg GPM MG430 4.2.1.11 enolase 2pg ↔ pep ENO MG407 2.7.1.40 pyruvate kinase pep + adp → pyr + atp PYK MG216 1.1.1.27 lactate dehydrogenase pyr + nadh ↔ lac + nad LDH MG460 1.1.1.94 sn-glycerol-3-phosphate dehydrogenase dhp + nadh → g3p + nad GPS n.i. 2.3.1.15 sn-glycerol-3-phosphate acyltransferase g3p + pal → mag PLSb n.i. 2.3.1.51 1-acyl-sn-glycerol-3-phosphate mag + pal → dag PLSc MG212 acyltransferase 2.7.7.41 phosphatidate cytidyltransferase dag + ctp → cdp-dag + pp CDS MG437 cdp-dag + ser → pser + 2.7.8.8 phosphatidylserine synthase PSS n.i. cmp 4.1.1.65 phosphatidylserine decarboxylase pser → peta PSD n.i. -
Q 297 Suppl USE
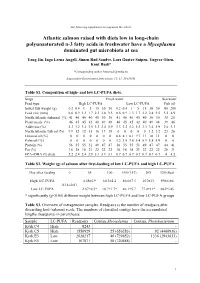
The following supplement accompanies the article Atlantic salmon raised with diets low in long-chain polyunsaturated n-3 fatty acids in freshwater have a Mycoplasma dominated gut microbiota at sea Yang Jin, Inga Leena Angell, Simen Rød Sandve, Lars Gustav Snipen, Yngvar Olsen, Knut Rudi* *Corresponding author: [email protected] Aquaculture Environment Interactions 11: 31–39 (2019) Table S1. Composition of high- and low LC-PUFA diets. Stage Fresh water Sea water Feed type High LC-PUFA Low LC-PUFA Fish oil Initial fish weight (g) 0.2 0.4 1 5 15 30 50 0.2 0.4 1 5 15 30 50 80 200 Feed size (mm) 0.6 0.9 1.3 1.7 2.2 2.8 3.5 0.6 0.9 1.3 1.7 2.2 2.8 3.5 3.5 4.9 North Atlantic fishmeal (%) 41 40 40 40 40 30 30 41 40 40 40 40 30 30 35 25 Plant meals (%) 46 45 45 42 40 49 48 46 45 45 42 40 49 48 39 46 Additives (%) 3.3 3.2 3.2 3.5 3.3 3.4 3.9 3.3 3.2 3.2 3.5 3.3 3.4 3.9 2.6 3.3 North Atlantic fish oil (%) 9.9 12 12 15 16 17 18 0 0 0 0 0 1.2 1.2 23 26 Linseed oil (%) 0 0 0 0 0 0 0 6.8 8.1 8.1 9.7 11 10 11 0 0 Palm oil (%) 0 0 0 0 0 0 0 3.2 3.8 3.8 5.4 5.9 5.8 5.9 0 0 Protein (%) 56 55 55 51 49 47 47 56 55 55 51 49 47 47 44 41 Fat (%) 16 18 18 21 22 22 22 16 18 18 21 22 22 22 28 31 EPA+DHA (% diet) 2.2 2.4 2.4 2.9 3.1 3.1 3.1 0.7 0.7 0.7 0.7 0.7 0.7 0.7 4 4.2 Table S2. -

Effects of High Forage/Concentrate Diet on Volatile Fatty Acid
animals Article Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen Lijun Wang 1,2, Guangning Zhang 2, Yang Li 2 and Yonggen Zhang 2,* 1 College of Animal Science and Technology, Qingdao Agricultural university, No. 700 of Changcheng Road, Qingdao 266000, China; [email protected] 2 College of Animal Science and Technology, Northeast Agricultural University, No. 600 of Changjiang Road, Harbin 150030, China; [email protected] (G.Z.); [email protected] (Y.L.) * Correspondence: [email protected]; Tel./Fax: +86-451-5519-0840 Received: 15 January 2020; Accepted: 28 January 2020; Published: 30 January 2020 Simple Summary: The rumen is well known as a natural bioreactor for highly efficient degradation of fibers, and rumen microbes play an important role on fiber degradation. Carbohydrates are fermented by a variety of bacteria in the rumen and transformed into volatile fatty acids (VFAs) by the corresponding enzymes. However, the content of forage in the diet affects the metabolism of cellulose degradation and VFA production. Therefore, we combine metabolism and metagenomics to explore the effects of High forage/concentrate diets and sampling time on enzymes and microorganisms involved in the metabolism of fiber and VFA in cow rumen. This study showed that propionate formation via the succinic pathway, in which succinate CoA synthetase (EC 6.2.1.5) and propionyl CoA carboxylase (EC 2.8.3.1) were key enzymes. Butyrate formation via the succinic pathway, in which phosphate butyryltransferase (EC 2.3.1.19), butyrate kinase (EC 2.7.2.7) and pyruvate ferredoxin oxidoreductase (EC 1.2.7.1) are the important enzymes. -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

Nondigestible Carbohydrates, Butyrate, and Butyrate-Producing Bacteria
Critical Reviews in Food Science and Nutrition ISSN: 1040-8398 (Print) 1549-7852 (Online) Journal homepage: https://www.tandfonline.com/loi/bfsn20 Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria Xiaodan Fu, Zhemin Liu, Changliang Zhu, Haijin Mou & Qing Kong To cite this article: Xiaodan Fu, Zhemin Liu, Changliang Zhu, Haijin Mou & Qing Kong (2018): Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria, Critical Reviews in Food Science and Nutrition, DOI: 10.1080/10408398.2018.1542587 To link to this article: https://doi.org/10.1080/10408398.2018.1542587 Published online: 22 Dec 2018. Submit your article to this journal Article views: 112 View Crossmark data Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=bfsn20 CRITICAL REVIEWS IN FOOD SCIENCE AND NUTRITION https://doi.org/10.1080/10408398.2018.1542587 REVIEW Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria Xiaodan Fu, Zhemin Liu, Changliang Zhu, Haijin Mou, and Qing Kong College of Food Science and Engineering, Ocean University of China, Qingdao, China ABSTRACT KEYWORDS Nondigestible carbohydrates (NDCs) are fermentation substrates in the colon after escaping diges- Nondigestible carbohy- tion in the upper gastrointestinal tract. Among NDCs, resistant starch is not hydrolyzed by pancre- drates; oligosaccharides; atic amylases but can be degraded by enzymes produced by large intestinal bacteria, including short-chain fatty acids; butyrate; butyrate- clostridia, bacteroides, and bifidobacteria. Nonstarch polysaccharides, such as pectin, guar gum, producing bacteria alginate, arabinoxylan, and inulin fructans, and nondigestible oligosaccharides and their deriva- tives, can also be fermented by beneficial bacteria in the large intestine. Butyrate is one of the most important metabolites produced through gastrointestinal microbial fermentation and func- tions as a major energy source for colonocytes by directly affecting the growth and differentiation of colonocytes. -

Phyre 2 Results for P50456
Email [email protected] Description P50456 Thu Jan 5 12:04:43 GMT Date 2012 Unique Job d92874c542ac9979 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:transcription Chain: A: PDB Molecule:transcriptional regulator, rok family; 1 c1z05A_ 100.0 45 Alignment PDBTitle: crystal structure of the rok family transcriptional regulator, homolog2 of e.coli mlc protein. PDB header:transcription 2 c1z6rC_ Alignment 100.0 98 Chain: C: PDB Molecule:mlc protein; PDBTitle: crystal structure of mlc from escherichia coli PDB header:transferase Chain: A: PDB Molecule:n-acetylglucosamine kinase; 3 c2hoeA_ 100.0 23 Alignment PDBTitle: crystal structure of n-acetylglucosamine kinase (tm1224) from2 thermotoga maritima at 2.46 a resolution PDB header:transferase Chain: A: PDB Molecule:glucokinase; 4 c3mcpA_ 100.0 18 Alignment PDBTitle: crystal structure of glucokinase (bdi_1628) from parabacteroides2 distasonis atcc 8503 at 3.00 a resolution PDB header:transferase Chain: D: PDB Molecule:glucokinase; 5 c2qm1D_ 100.0 24 Alignment PDBTitle: crystal structure of glucokinase from enterococcus faecalis PDB header:transferase Chain: B: PDB Molecule:glucokinase; 6 c3vgkB_ 100.0 25 Alignment PDBTitle: crystal structure of a rok family glucokinase from streptomyces2 griseus PDB header:transferase Chain: A: PDB Molecule:hypothetical sugar kinase; 7 c3r8eA_ Alignment 100.0 19 PDBTitle: crystal structure of a hypothetical sugar kinase (chu_1875) from2 cytophaga hutchinsonii atcc 33406 at 1.65 -

Roles of the Methylcitrate and Methylmalonyl-COA Pathways in Mycobacterial Metabolism and Pathogenesis" (2012)
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2012 Roles of the Methylcitrate and Methylmalonyl- COA Pathways in Mycobacterial Metabolism and Pathogenesis Manisha Ulhas Lotlikar Follow this and additional works at: http://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons Recommended Citation Lotlikar, Manisha Ulhas, "Roles of the Methylcitrate and Methylmalonyl-COA Pathways in Mycobacterial Metabolism and Pathogenesis" (2012). Student Theses and Dissertations. Paper 245. This Thesis is brought to you for free and open access by Digital Commons @ RU. It has been accepted for inclusion in Student Theses and Dissertations by an authorized administrator of Digital Commons @ RU. For more information, please contact [email protected]. ROLES OF THE METHYLCITRATE AND METHYLMALONYL-COA PATHWAYS IN MYCOBACTERIAL METABOLISM AND PATHOGENESIS A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Manisha Ulhas Lotlikar June 2012 © Copyright by Manisha Ulhas Lotlikar 2012 ROLES OF THE METHYLCITRATE AND METHYLMALONYL-COA PATHWAYS IN MYCOBACTERIAL METABOLISM AND PATHOGENESIS Manisha Ulhas Lotlikar, Ph.D. The Rockefeller University 2012 Mycobacterium tuberculosis has been a human pathogen for the history of mankind, but we are only now beginning to understand how it is able to survive and persist indefinitely in the host. Understanding carbon metabolism of the pathogen during infection is key, not only as a source of potential drug targets, but also for elucidating the environment in vivo, so that drugs can be tested under relevant conditions. Studies have revealed that, during infection, M. tuberculosis relies on gluconeogenic carbon sources rather than sugars.