Dissecting and Reprogramming the Folding and Assembly of Tandem-Repeat Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ankrd9 Is a Metabolically-Controlled Regulator of Impdh2 Abundance and Macro-Assembly
ANKRD9 IS A METABOLICALLY-CONTROLLED REGULATOR OF IMPDH2 ABUNDANCE AND MACRO-ASSEMBLY by Dawn Hayward A dissertation submitted to The Johns Hopkins University in conformity with the requirements of the degree of Doctor of Philosophy Baltimore, Maryland April 2019 ABSTRACT Members of a large family of Ankyrin Repeat Domains proteins (ANKRD) regulate numerous cellular processes by binding and changing properties of specific protein targets. We show that interactions with a target protein and the functional outcomes can be markedly altered by cells’ metabolic state. ANKRD9 facilitates degradation of inosine monophosphate dehydrogenase 2 (IMPDH2), the rate-limiting enzyme in GTP biosynthesis. Under basal conditions ANKRD9 is largely segregated from the cytosolic IMPDH2 by binding to vesicles. Upon nutrient limitation, ANKRD9 loses association with vesicles and assembles with IMPDH2 into rod-like structures, in which IMPDH2 is stable. Inhibition of IMPDH2 with Ribavirin favors ANKRD9 binding to rods. The IMPDH2/ANKRD9 assembly is reversed by guanosine, which restores association of ANKRD9 with vesicles. The conserved Cys109Cys110 motif in ANKRD9 is required for the vesicles-to-rods transition as well as binding and regulation of IMPDH2. ANKRD9 knockdown increases IMPDH2 levels and prevents formation of IMPDH2 rods upon nutrient limitation. Thus, the status of guanosine pools affects the mode of ANKRD9 action towards IMPDH2. Advisor: Dr. Svetlana Lutsenko, Department of Physiology, Johns Hopkins University School of Medicine Second reader: -

Repeat Proteins Challenge the Concept of Structural Domains
844 Biochemical Society Transactions (2015) Volume 43, part 5 Repeat proteins challenge the concept of structural domains Rocıo´ Espada*1, R. Gonzalo Parra*1, Manfred J. Sippl†, Thierry Mora‡, Aleksandra M. Walczak§ and Diego U. Ferreiro*2 *Protein Physiology Lab, Dep de Qu´ımica Biologica, ´ Facultad de Ciencias Exactas y Naturales, UBA-CONICET-IQUIBICEN, Buenos Aires, C1430EGA, Argentina †Center of Applied Molecular Engineering, Division of Bioinformatics, Department of Molecular Biology, University of Salzburg, 5020 Salzburg, Austria ‡Laboratoire de physique statistique, CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France §Laboratoire de physique theorique, ´ CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France Abstract Structural domains are believed to be modules within proteins that can fold and function independently. Some proteins show tandem repetitions of apparent modular structure that do not fold independently, but rather co-operate in stabilizing structural forms that comprise several repeat-units. For many natural repeat-proteins, it has been shown that weak energetic links between repeats lead to the breakdown of co-operativity and the appearance of folding sub-domains within an apparently regular repeat array. The quasi-1D architecture of repeat-proteins is crucial in detailing how the local energetic balances can modulate the folding dynamics of these proteins, which can be related to the physiological behaviour of these ubiquitous biological systems. Introduction and between repeats, challenging the concept of structural It was early on noted that many natural proteins typically domain. collapse stretches of amino acid chains into compact units, defining structural domains [1]. These domains typically correlate with biological activities and many modern proteins can be described as composed by novel ‘domain arrange- Definition of the repeat-units ments’ [2]. -

A Structural Guide to Proteins of the NF-Kb Signaling Module
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A Structural Guide to Proteins of the NF-kB Signaling Module Tom Huxford1 and Gourisankar Ghosh2 1Department of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182-1030 2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0375 Correspondence: [email protected] The prosurvival transcription factor NF-kB specifically binds promoter DNA to activate target gene expression. NF-kB is regulated through interactions with IkB inhibitor proteins. Active proteolysis of these IkB proteins is, in turn, under the control of the IkB kinase complex (IKK). Together, these three molecules form the NF-kB signaling module. Studies aimed at charac- terizing the molecular mechanisms of NF-kB, IkB, and IKK in terms of their three-dimen- sional structures have lead to a greater understanding of this vital transcription factor system. F-kB is a master transcription factor that from the perspective of their three-dimensional Nresponds to diverse cell stimuli by activat- structures. ing the expression of stress response genes. Multiple signals, including cytokines, growth factors, engagement of the T-cell receptor, and NF-kB bacterial and viral products, induce NF-kB Introduction to NF-kB transcriptional activity (Hayden and Ghosh 2008). A point of convergence for the myriad NF-kB was discovered in the laboratory of of NF-kB inducing signals is the IkB kinase David Baltimore as a nuclear activity with bind- complex (IKK). Active IKK in turn controls ing specificity toward a ten-base-pair DNA transcription factor NF-kB by regulating pro- sequence 50-GGGACTTTCC-30 present within teolysis of the IkB inhibitor protein (Fig. -
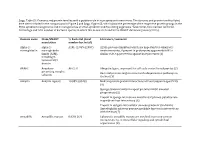
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Eukaryotic Genome Annotation
Comparative Features of Multicellular Eukaryotic Genomes (2017) (First three statistics from www.ensembl.org; other from original papers) C. elegans A. thaliana D. melanogaster M. musculus H. sapiens Species name Nematode Thale Cress Fruit Fly Mouse Human Size (Mb) 103 136 143 3,482 3,555 # Protein-coding genes 20,362 27,655 13,918 22,598 20,338 (25,498 (13,601 original (30,000 (30,000 original est.) original est.) original est.) est.) Transcripts 58,941 55,157 34,749 131,195 200,310 Gene density (#/kb) 1/5 1/4.5 1/8.8 1/83 1/97 LINE/SINE (%) 0.4 0.5 0.7 27.4 33.6 LTR (%) 0.0 4.8 1.5 9.9 8.6 DNA Elements 5.3 5.1 0.7 0.9 3.1 Total repeats 6.5 10.5 3.1 38.6 46.4 Exons % genome size 27 28.8 24.0 per gene 4.0 5.4 4.1 8.4 8.7 average size (bp) 250 506 Introns % genome size 15.6 average size (bp) 168 Arabidopsis Chromosome Structures Sorghum Whole Genome Details Characterizing the Proteome The Protein World • Sequencing has defined o Many, many proteins • How can we use this data to: o Define genes in new genomes o Look for evolutionarily related genes o Follow evolution of genes ▪ Mixing of domains to create new proteins o Uncover important subsets of genes that ▪ That deep phylogenies • Plants vs. animals • Placental vs. non-placental animals • Monocots vs. dicots plants • Common nomenclature needed o Ensure consistency of interpretations InterPro (http://www.ebi.ac.uk/interpro/) Classification of Protein Families • Intergrated documentation resource for protein super families, families, domains and functional sites o Mitchell AL, Attwood TK, Babbitt PC, et al. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

WSB1: from Homeostasis to Hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew Macisaac1,2,3 and Douglas James Demetrick1,2,3,4*
Haque et al. Journal of Biomedical Science (2016) 23:61 DOI 10.1186/s12929-016-0270-3 REVIEW Open Access WSB1: from homeostasis to hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew MacIsaac1,2,3 and Douglas James Demetrick1,2,3,4* Abstract The wsb1 gene has been identified to be important in developmental biology and cancer. A complex transcriptional regulation of wsb1 yields at least three functional transcripts. The major expressed isoform, WSB1 protein, is a substrate recognition protein within an E3 ubiquitin ligase, with the capability to bind diverse targets and mediate ubiquitinylation and proteolytic degradation. Recent data suggests a new role for WSB1 as a component of a neuroprotective pathway which results in modification and aggregation of neurotoxic proteins such as LRRK2 in Parkinson’s Disease, via an unusual mode of protein ubiquitinylation. WSB1 is also involved in thyroid hormone homeostasis, immune regulation and cellular metabolism, particularly glucose metabolism and hypoxia. In hypoxia, wsb1 is a HIF-1 target, and is a regulator of the degradation of diverse proteins associated with the cellular response to hypoxia, including HIPK2, RhoGDI2 and VHL. Major roles are to both protect HIF-1 function through degradation of VHL, and decrease apoptosis through degradation of HIPK2. These activities suggest a role for wsb1 in cancer cell proliferation and metastasis. As well, recent work has identified a role for WSB1 in glucose metabolism, and perhaps in mediating the Warburg effect in cancer cells by maintaining the function of HIF1. Furthermore, studies of cancer specimens have identified dysregulation of wsb1 associated with several types of cancer, suggesting a biologically relevant role in cancer development and/or progression. -

An Unusual Hydrophobic Core Confers Extreme Flexibility to HEAT Repeat Proteins
1596 Biophysical Journal Volume 99 September 2010 1596–1603 An Unusual Hydrophobic Core Confers Extreme Flexibility to HEAT Repeat Proteins Christian Kappel, Ulrich Zachariae, Nicole Do¨lker, and Helmut Grubmu¨ller* Department of Theoretical and Computational Biophysics, Max Planck Institute for Biophysical Chemistry, Go¨ttingen, Germany ABSTRACT Alpha-solenoid proteins are suggested to constitute highly flexible macromolecules, whose structural variability and large surface area is instrumental in many important protein-protein binding processes. By equilibrium and nonequilibrium molecular dynamics simulations, we show that importin-b, an archetypical a-solenoid, displays unprecedentedly large and fully reversible elasticity. Our stretching molecular dynamics simulations reveal full elasticity over up to twofold end-to-end extensions compared to its bound state. Despite the absence of any long-range intramolecular contacts, the protein can return to its equilibrium structure to within 3 A˚ backbone RMSD after the release of mechanical stress. We find that this extreme degree of flexibility is based on an unusually flexible hydrophobic core that differs substantially from that of structurally similar but more rigid globular proteins. In that respect, the core of importin-b resembles molten globules. The elastic behavior is dominated by nonpolar interactions between HEAT repeats, combined with conformational entropic effects. Our results suggest that a-sole- noid structures such as importin-b may bridge the molecular gap between completely structured and intrinsically disordered proteins. INTRODUCTION Solenoid proteins, consisting of repeating arrays of simple on repeat proteins focus on the folding and unfolding basic structural motifs, account for >5% of the genome of mechanism (14–16), an atomic force microscopy study on multicellular organisms (1). -

Supporting Information
Supporting Information Barquilla et al. 10.1073/pnas.0802668105 SI Materials and Methods The purification of recombinant GST proteins using glutathione Bioinformatics, Plasmids Constructs, and Cell Lines. Searches for T. Sepharose 4B (Amersham) was performed according to the brucei TOR, raptor, AVO3, and FKBP12 orthologues were manufacturer’s instructions and the proteins were dialyzed performed using GeneDB (www.genedb.org) and NCBI Blast against PBS. (www.ncbi.nlm.nih.gov/BLAST/). Motif analyses were carried out using InterPro and Prosite. Antibodies. Mouse anti-TbFKBP12 monoclonal antibodies and GeneDB database accession numbers of these orthologues rabbit polyclonal anti-TOR antibodies were produced by inject- are: TbTOR1 (Tb10.6k15.2060), TbTOR2 (Tb927.4.420), ing the recombinant proteins into a BALB-C mouse or rabbit TbTOR-like 1 (Tb927.4.800), TbTOR-like 2 (Tb927.1.1930), using standard immunization protocols. Experiments involving TbRaptor (Tb11.03.0460), TbAVO3 (Tb927.8.3200), animals were approved by the National Animal Research Com- TbFKBP12 (Tb927.7.3420), and TbRaptor-like (Tb927.1.2190). mittee. Anti-TbFKBP12 antibodies were raised against the DNA fragments (400–700 bp) of the TbTOR1, TbTOR2, whole protein fused to GST, and hybridomes F12–3C7H11 and TbTOR-like 1, TbRaptor, TbAVO3, TbFKBP12, and TbRaptor- F12–7D10 H8 were selected by standard monoclonal selection like were amplified by PCR using pairs of specific primers (Table procedures as anti-TbFKBP12 monoclonal antibody producers. S1) and cloned into pGEM-T (Promega). All constructs were Anti-TbTOR antibodies were raised against nonhomologous sequenced to confirm that the subcloned DNA fragments had no TbTOR CЈ-terminal regions between the kinase domain and the mutations. -

Structural and Biochemical Consequences of Disease-Causing Mutations in the Ankyrin Repeat Domain of the Human TRPV4 Channel
Structural and Biochemical Consequences of Disease-Causing Mutations in the Ankyrin Repeat Domain of the Human TRPV4 Channel The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Inada, Hitoshi, Erik Procko, Marcos Sotomayor, and Rachelle Gaudet. 2012. Structural and biochemical consequences of disease- causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 51(31): 6195-6206. Published Version doi:10.1021/bi300279b Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:11688820 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel Hitoshi Inada†, Erik Procko†,‡, Marcos Sotomayor†,§, and Rachelle Gaudet†,* †Department of Molecular and Cellular Biology, Harvard University, 52 Oxford Street, Cambridge, MA 02138 ‡Present address: Howard Hughes Medical Institute and Department of Biochemistry, University of Washington, Seattle, WA 98195, USA §Howard Hughes Medical Institute and Department of Neurobiology, Harvard Medical School, Boston, MA 02115, USA AUTHOR EMAIL ADDRESS: [email protected] TITLE RUNNING HEAD: Structural analysis of human TRPV4-ARD CORRESPONDING AUTHOR FOOTNOTE: *To whom correspondence should be addressed: Rachelle Gaudet, Department of Molecular and Cellular Biology, Harvard University, 52 Oxford Street, Cambridge, MA 02138, USA, Tel.: (617) 495-5616; Fax: (617) 496-9684; E-mail: [email protected] FOOTNOTES: This work was funded by NIH grant R01GM081340 to RG. -

'Design of Armadillo Repeat Protein Scaffolds'
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2008 Design of armadillo repeat protein scaffolds Parmeggiani, F Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-4818 Dissertation Published Version Originally published at: Parmeggiani, F. Design of armadillo repeat protein scaffolds. 2008, University of Zurich, Faculty of Medicine. Design of Armadillo Repeat Protein Scaffolds Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr. sc. nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Fabio Parmeggiani aus Italien Promotionskomitee Prof. Dr. Andreas Plückthun (Leitung der Dissertation) Prof. Dr. Markus Grütter Prof. Dr. Donald Hilvert Zürich, 2008 To my grandfather, who showed me the essence of curiosity “It is easy to add more of the same to old knowledge, but it is difficult to explain the new” Benno Müller-Hill Table of Contents 1 Table of contents Summary 3 Chapter 1: Peptide binders 7 Part I: Peptide binders 8 Modular interaction domains as natural peptide binders 9 SH2 domains 10 PTB domains 10 PDZ domains 12 14-3-3 domains 12 SH3 domains 12 WW domains 12 Peptide recognition by major histocompatibility complexes 13 Repeat proteins targeting peptides 15 Armadillo repeat proteins 16 Tetratricopeptide repeat proteins (TPR) 16 Beta propellers 16 Peptide binding by antibodies 18 Alternative scaffolds: a new approach to peptide binding 20 Part -

Take Home Lessons from Studies of Related Proteins
Available online at www.sciencedirect.com Take home lessons from studies of related proteins * Adrian A Nickson, Beth G Wensley and Jane Clarke The ‘Fold Approach’ involves a detailed analysis of the folding The malleability of protein folding pathways of several topologically, structurally and/or evolutionarily A unifying folding mechanism related proteins. Such studies can reveal determinants of the In the early days of the ‘protein-folding problem’, three folding mechanism beyond the gross topology, and can dissect competing mechanisms were proposed that described the residues required for folding from those required for stability how a polypeptide chain might fold to the native state: or function. While this approach has not yet matured to the nucleation [3], hydrophobic-collapse [4] and diffusion- point where we can predict the native conformation of any collision (framework) [5]. However, an early F-value polypeptide chain in silico, it has been able to highlight, analysis of the small protein chymotrypsin inhibitor 2 amongst others, the specific residues that are responsible for (CI2) demonstrated that none of these mechanisms was nucleation, pathway malleability, kinetic intermediates, chain appropriate, since secondary and tertiary structure formed knotting, internal friction and Paracelsus switches. Some of the concomitantly [6]. Thus the nucleation-condensation most interesting discoveries have resulted from the attempt to mechanism was introduced [7], in which long-range con- explain differences between homologues. tacts set up the initial topology of the protein (incurring a Address substantial entropic loss with minimal enthalpic gain), Department of Chemistry, University of Cambridge, Lensfield Rd, followed by a rapid collapse to the native state (with Cambridge CB2 1EW, UK minimal entropic loss but substantial enthalpic gain).