Repeat Proteins Challenge the Concept of Structural Domains
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ankrd9 Is a Metabolically-Controlled Regulator of Impdh2 Abundance and Macro-Assembly
ANKRD9 IS A METABOLICALLY-CONTROLLED REGULATOR OF IMPDH2 ABUNDANCE AND MACRO-ASSEMBLY by Dawn Hayward A dissertation submitted to The Johns Hopkins University in conformity with the requirements of the degree of Doctor of Philosophy Baltimore, Maryland April 2019 ABSTRACT Members of a large family of Ankyrin Repeat Domains proteins (ANKRD) regulate numerous cellular processes by binding and changing properties of specific protein targets. We show that interactions with a target protein and the functional outcomes can be markedly altered by cells’ metabolic state. ANKRD9 facilitates degradation of inosine monophosphate dehydrogenase 2 (IMPDH2), the rate-limiting enzyme in GTP biosynthesis. Under basal conditions ANKRD9 is largely segregated from the cytosolic IMPDH2 by binding to vesicles. Upon nutrient limitation, ANKRD9 loses association with vesicles and assembles with IMPDH2 into rod-like structures, in which IMPDH2 is stable. Inhibition of IMPDH2 with Ribavirin favors ANKRD9 binding to rods. The IMPDH2/ANKRD9 assembly is reversed by guanosine, which restores association of ANKRD9 with vesicles. The conserved Cys109Cys110 motif in ANKRD9 is required for the vesicles-to-rods transition as well as binding and regulation of IMPDH2. ANKRD9 knockdown increases IMPDH2 levels and prevents formation of IMPDH2 rods upon nutrient limitation. Thus, the status of guanosine pools affects the mode of ANKRD9 action towards IMPDH2. Advisor: Dr. Svetlana Lutsenko, Department of Physiology, Johns Hopkins University School of Medicine Second reader: -

A Structural Guide to Proteins of the NF-Kb Signaling Module
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A Structural Guide to Proteins of the NF-kB Signaling Module Tom Huxford1 and Gourisankar Ghosh2 1Department of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182-1030 2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0375 Correspondence: [email protected] The prosurvival transcription factor NF-kB specifically binds promoter DNA to activate target gene expression. NF-kB is regulated through interactions with IkB inhibitor proteins. Active proteolysis of these IkB proteins is, in turn, under the control of the IkB kinase complex (IKK). Together, these three molecules form the NF-kB signaling module. Studies aimed at charac- terizing the molecular mechanisms of NF-kB, IkB, and IKK in terms of their three-dimen- sional structures have lead to a greater understanding of this vital transcription factor system. F-kB is a master transcription factor that from the perspective of their three-dimensional Nresponds to diverse cell stimuli by activat- structures. ing the expression of stress response genes. Multiple signals, including cytokines, growth factors, engagement of the T-cell receptor, and NF-kB bacterial and viral products, induce NF-kB Introduction to NF-kB transcriptional activity (Hayden and Ghosh 2008). A point of convergence for the myriad NF-kB was discovered in the laboratory of of NF-kB inducing signals is the IkB kinase David Baltimore as a nuclear activity with bind- complex (IKK). Active IKK in turn controls ing specificity toward a ten-base-pair DNA transcription factor NF-kB by regulating pro- sequence 50-GGGACTTTCC-30 present within teolysis of the IkB inhibitor protein (Fig. -
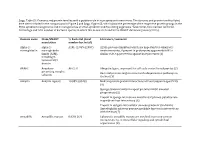
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

WSB1: from Homeostasis to Hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew Macisaac1,2,3 and Douglas James Demetrick1,2,3,4*
Haque et al. Journal of Biomedical Science (2016) 23:61 DOI 10.1186/s12929-016-0270-3 REVIEW Open Access WSB1: from homeostasis to hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew MacIsaac1,2,3 and Douglas James Demetrick1,2,3,4* Abstract The wsb1 gene has been identified to be important in developmental biology and cancer. A complex transcriptional regulation of wsb1 yields at least three functional transcripts. The major expressed isoform, WSB1 protein, is a substrate recognition protein within an E3 ubiquitin ligase, with the capability to bind diverse targets and mediate ubiquitinylation and proteolytic degradation. Recent data suggests a new role for WSB1 as a component of a neuroprotective pathway which results in modification and aggregation of neurotoxic proteins such as LRRK2 in Parkinson’s Disease, via an unusual mode of protein ubiquitinylation. WSB1 is also involved in thyroid hormone homeostasis, immune regulation and cellular metabolism, particularly glucose metabolism and hypoxia. In hypoxia, wsb1 is a HIF-1 target, and is a regulator of the degradation of diverse proteins associated with the cellular response to hypoxia, including HIPK2, RhoGDI2 and VHL. Major roles are to both protect HIF-1 function through degradation of VHL, and decrease apoptosis through degradation of HIPK2. These activities suggest a role for wsb1 in cancer cell proliferation and metastasis. As well, recent work has identified a role for WSB1 in glucose metabolism, and perhaps in mediating the Warburg effect in cancer cells by maintaining the function of HIF1. Furthermore, studies of cancer specimens have identified dysregulation of wsb1 associated with several types of cancer, suggesting a biologically relevant role in cancer development and/or progression. -

Structural and Biochemical Consequences of Disease-Causing Mutations in the Ankyrin Repeat Domain of the Human TRPV4 Channel
Structural and Biochemical Consequences of Disease-Causing Mutations in the Ankyrin Repeat Domain of the Human TRPV4 Channel The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Inada, Hitoshi, Erik Procko, Marcos Sotomayor, and Rachelle Gaudet. 2012. Structural and biochemical consequences of disease- causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 51(31): 6195-6206. Published Version doi:10.1021/bi300279b Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:11688820 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel Hitoshi Inada†, Erik Procko†,‡, Marcos Sotomayor†,§, and Rachelle Gaudet†,* †Department of Molecular and Cellular Biology, Harvard University, 52 Oxford Street, Cambridge, MA 02138 ‡Present address: Howard Hughes Medical Institute and Department of Biochemistry, University of Washington, Seattle, WA 98195, USA §Howard Hughes Medical Institute and Department of Neurobiology, Harvard Medical School, Boston, MA 02115, USA AUTHOR EMAIL ADDRESS: [email protected] TITLE RUNNING HEAD: Structural analysis of human TRPV4-ARD CORRESPONDING AUTHOR FOOTNOTE: *To whom correspondence should be addressed: Rachelle Gaudet, Department of Molecular and Cellular Biology, Harvard University, 52 Oxford Street, Cambridge, MA 02138, USA, Tel.: (617) 495-5616; Fax: (617) 496-9684; E-mail: [email protected] FOOTNOTES: This work was funded by NIH grant R01GM081340 to RG. -

Dissecting and Reprogramming the Folding and Assembly of Tandem-Repeat Proteins
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Apollo Dissecting and reprogramming the folding and assembly of tandem-repeat proteins Pamela J.E. Rowling1, Elin M. Sivertsson1, Albert Perez-Riba1, Ewan R. G. Main2*, Laura S. Itzhaki1* 1University of Cambridge Department of Pharmacology, Tennis Court Road, Cambridge CB2 1PD, UK. 2School of Biological and Chemical Sciences, Queen Mary, University of London, London E1 4NS, UK. *To whom correspondence should be addressed: [email protected] and [email protected] Studying protein folding and protein design in globular proteins presents significant challenges because of the two related features, topological complexity and cooperativity. In contrast, tandem-repeat proteins have regular and modular structures composed of linearly arrayed motifs. This means that the biophysics of even giant repeat proteins is highly amenable to dissection and to rational design. Here we discuss what has been learnt about the folding mechanisms of tandem- repeat proteins. The defining features that have emerged are: (i) accessibility of multiple distinct routes between denatured and native states, both at equilibrium and under kinetic conditions; (ii) different routes are favoured for folding versus unfolding; (iii) unfolding energy barriers are broad, reflecting stepwise unraveling of an array repeat by repeat; (iv) highly cooperative unfolding at equilibrium and the potential for exceptionally high thermodynamic stabilities by introducing consensus residues; (v) under force, helical-repeat structures are very weak with non-cooperative unfolding leading to elasticity and buffering effects. This level of understanding should enable us to create repeat proteins with made-to-measure folding mechanisms, in which one can dial into the sequence the order of repeat folding, number of pathways taken, step size (cooperativity) and fine-structure of the kinetic energy barriers. -

Take Home Lessons from Studies of Related Proteins
Available online at www.sciencedirect.com Take home lessons from studies of related proteins * Adrian A Nickson, Beth G Wensley and Jane Clarke The ‘Fold Approach’ involves a detailed analysis of the folding The malleability of protein folding pathways of several topologically, structurally and/or evolutionarily A unifying folding mechanism related proteins. Such studies can reveal determinants of the In the early days of the ‘protein-folding problem’, three folding mechanism beyond the gross topology, and can dissect competing mechanisms were proposed that described the residues required for folding from those required for stability how a polypeptide chain might fold to the native state: or function. While this approach has not yet matured to the nucleation [3], hydrophobic-collapse [4] and diffusion- point where we can predict the native conformation of any collision (framework) [5]. However, an early F-value polypeptide chain in silico, it has been able to highlight, analysis of the small protein chymotrypsin inhibitor 2 amongst others, the specific residues that are responsible for (CI2) demonstrated that none of these mechanisms was nucleation, pathway malleability, kinetic intermediates, chain appropriate, since secondary and tertiary structure formed knotting, internal friction and Paracelsus switches. Some of the concomitantly [6]. Thus the nucleation-condensation most interesting discoveries have resulted from the attempt to mechanism was introduced [7], in which long-range con- explain differences between homologues. tacts set up the initial topology of the protein (incurring a Address substantial entropic loss with minimal enthalpic gain), Department of Chemistry, University of Cambridge, Lensfield Rd, followed by a rapid collapse to the native state (with Cambridge CB2 1EW, UK minimal entropic loss but substantial enthalpic gain). -

(TRP) Cation Channels Could Explain Smell, Taste, And/Or Chemesthesis Disorders
Interactions of SARS-CoV-2 spike protein and transient receptor potential (TRP) cation channels could explain smell, taste, and/or chemesthesis disorders Halim Maaroufi Institut de biologie intégrative et des systèmes (IBIS). Université Laval. Quebec city, Quebec, Canada. [email protected] IN BRIEF SARS-CoV-2 S protein contains two ankyrin repeat binding motifs (S-ARBMs) that could interact with ankyrin repeat domains (ARDs) of transient receptor potential (TRP) channels. The latter play a role in olfaction, taste, and/or chemesthesis (OTC), suggesting that their dysfunction by S protein could explain OTC disorders in COVID-19 disease. Pharmacological manipulation of TRPs-ARDs could be used for prophylactic or treatments in SARS-CoV-2 infections. HIGHLIGHTS SARS-CoV-2 infection is associated to olfactory, taste, and/or chemesthesis (OTC) disorders (OTCD). Transient receptor potential (TRP) channels play a role in OTC. S protein contains ARBMs that could interact with ARDs of TRPs, thus inducing OTCD. TRPs ligands as possible preventive treatments against COVID-19. KEYWORDS Loss, Olfactory, Taste, Chemesthesis, SARS-CoV-2, S protein, Ankyrin repeat, Ankyrin repeat binding motif, TRPA1, TRPCs, TRPVs, TRPs ligands, DARPins ABBREVIATIONS SARS-CoV-2: severe acute respiratory syndrome coronavirus 2 COVID-19: Coronavirus disease-2019 S protein: Spike protein 1 RBD: Receptor binding domain HR1: Heptad repeat 1 OTC: olfactory, taste and chemesthesis OTCD: olfactory, taste and/or chemesthesis disorders TRP: Transient receptor potential -

The Contribution of the Ankyrin Repeat Domain of TRPV1 As a Thermal
bioRxiv preprint doi: https://doi.org/10.1101/641803; this version posted May 19, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 The contribution of the ankyrin repeat 2 domain of TRPV1 as a thermal module 3 4 E. Ladrón-de-Guevara 5 G. E. Rangel-Yescas 6 D. A. Fernández-Velasco 7 A. Torres-Larios 8 T. Rosenbaum 9 L. D. Islas 10 11 Abstract 12 The TRPV1 cation non-selective ion channel plays an essential role in thermosensation and perception of 13 other noxious stimuli. TRPV1 can be activated by low pH, high temperature or naturally occurring 14 pungent molecules such as allicin, capsaicin or resiniferatoxin. Its noxious thermal sensitivity makes it an 15 important participant as a thermal sensor in mammals. However, details of the mechanism of channel 16 activation by increases in temperature remain unclear. Here we used a combination of approaches to try 17 to understand the role of the ankyrin repeat domain (ARD) in channel behavior. First, a computational 18 modeling approach by coarse-grained molecular dynamics simulation of the whole TRPV1 embedded in 19 a phosphatidylcholine (POPC) and phosphatidylethanolamine (POPE) membrane provides insight into 20 the dynamics of this channel domain. Global analysis of the structural ensemble shows that the ankyrin 21 repeat domain is a region that sustains high fluctuations during dynamics at different temperatures. We 22 then performed biochemical and thermal stability studies of the purified ARD by means of circular 23 dichroism and tryptophan fluorescence and demonstrate that this region undergoes structural changes 24 at similar temperatures that lead to TRPV1 activation. -

Use of Protein Engineering Techniques to Elucidate Protein Folding Pathways
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book Progress in Molecular Biology and Translational Science, Vol. 84, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: Anna L. Mallam and Sophie E. Jackson, Use of Protein Engineering Techniques to Elucidate Protein Folding Pathways. In P. Michael Conn, editor: Progress in Molecular Biology and Translational Science, Vol. 84, Burlington: Academic Press, 2008, pp. 57-113. ISBN: 978-0-12-374595-8 © Copyright 2008 Elsevier Inc. Academic Press. Author's personal copy Use of Protein Engineering Techniques to Elucidate Protein Folding Pathways Anna L. Mallam and Sophie E. Jackson Department of Chemistry, Cambridge, CB2 1EW, United Kingdom I. Introduction............................................................................... 58 II. Early Protein Engineering Studies of Folding Pathways ........................ 59 III. Single Point Mutations and F‐Value Analysis...................................... 61 A. Mixed a/b Proteins .................................................................. 65 B. All‐a‐Helical Proteins............................................................... 69 C. All‐b‐Proteins........................................................................ -

S41467-020-15139-6.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-15139-6 OPEN An ankyrin-repeat and WRKY-domain-containing immune receptor confers stripe rust resistance in wheat ✉ Huan Wang 1,2, Shenghao Zou1, Yiwen Li 2, Fanyun Lin 2 & Dingzhong Tang1 Perception of pathogenic effectors in plants often relies on nucleotide-binding domain (NBS) and leucine-rich-repeat-containing (NLR) proteins. Some NLRs contain additional domains 1234567890():,; that function as integrated decoys for pathogen effector targets and activation of immune signalling. Wheat stripe rust is one of the most devastating diseases of crop plants. Here, we report the cloning of YrU1, a stripe rust resistance gene from the diploid wheat Triticum urartu, the progenitor of the A genome of hexaploid wheat. YrU1 encodes a coiled-coil-NBS-leucine- rich repeat protein with N-terminal ankyrin-repeat and C-terminal WRKY domains, repre- senting a unique NLR structure in plants. Database searches identify similar architecture only in wheat relatives. Transient expression of YrU1 in Nicotiana benthamiana does not induce cell death in the absence of pathogens. The ankyrin-repeat and coiled-coil domains of YrU1 self- associate, suggesting that homodimerisation is critical for YrU1 function. The identification and cloning of this disease resistance gene sheds light on NLR protein function and may facilitate breeding to control the devastating wheat stripe rust disease. 1 State Key Laboratory of Ecological Control of Fujian-Taiwan Crop Pests, Key Laboratory of Ministry of Education for Genetics, Breeding and Multiple Utilization of Crops, Plant Immunity Center, Fujian Agriculture and Forestry University, Fuzhou 350002, China. 2 State Key Laboratory of Plant Cell and Chromosome ✉ Engineering, Institute of Genetics and Development Biology, Chinese Academy of Sciences, Beijing 100101, China.