Use of Protein Engineering Techniques to Elucidate Protein Folding Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ankrd9 Is a Metabolically-Controlled Regulator of Impdh2 Abundance and Macro-Assembly
ANKRD9 IS A METABOLICALLY-CONTROLLED REGULATOR OF IMPDH2 ABUNDANCE AND MACRO-ASSEMBLY by Dawn Hayward A dissertation submitted to The Johns Hopkins University in conformity with the requirements of the degree of Doctor of Philosophy Baltimore, Maryland April 2019 ABSTRACT Members of a large family of Ankyrin Repeat Domains proteins (ANKRD) regulate numerous cellular processes by binding and changing properties of specific protein targets. We show that interactions with a target protein and the functional outcomes can be markedly altered by cells’ metabolic state. ANKRD9 facilitates degradation of inosine monophosphate dehydrogenase 2 (IMPDH2), the rate-limiting enzyme in GTP biosynthesis. Under basal conditions ANKRD9 is largely segregated from the cytosolic IMPDH2 by binding to vesicles. Upon nutrient limitation, ANKRD9 loses association with vesicles and assembles with IMPDH2 into rod-like structures, in which IMPDH2 is stable. Inhibition of IMPDH2 with Ribavirin favors ANKRD9 binding to rods. The IMPDH2/ANKRD9 assembly is reversed by guanosine, which restores association of ANKRD9 with vesicles. The conserved Cys109Cys110 motif in ANKRD9 is required for the vesicles-to-rods transition as well as binding and regulation of IMPDH2. ANKRD9 knockdown increases IMPDH2 levels and prevents formation of IMPDH2 rods upon nutrient limitation. Thus, the status of guanosine pools affects the mode of ANKRD9 action towards IMPDH2. Advisor: Dr. Svetlana Lutsenko, Department of Physiology, Johns Hopkins University School of Medicine Second reader: -

Repeat Proteins Challenge the Concept of Structural Domains
844 Biochemical Society Transactions (2015) Volume 43, part 5 Repeat proteins challenge the concept of structural domains Rocıo´ Espada*1, R. Gonzalo Parra*1, Manfred J. Sippl†, Thierry Mora‡, Aleksandra M. Walczak§ and Diego U. Ferreiro*2 *Protein Physiology Lab, Dep de Qu´ımica Biologica, ´ Facultad de Ciencias Exactas y Naturales, UBA-CONICET-IQUIBICEN, Buenos Aires, C1430EGA, Argentina †Center of Applied Molecular Engineering, Division of Bioinformatics, Department of Molecular Biology, University of Salzburg, 5020 Salzburg, Austria ‡Laboratoire de physique statistique, CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France §Laboratoire de physique theorique, ´ CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France Abstract Structural domains are believed to be modules within proteins that can fold and function independently. Some proteins show tandem repetitions of apparent modular structure that do not fold independently, but rather co-operate in stabilizing structural forms that comprise several repeat-units. For many natural repeat-proteins, it has been shown that weak energetic links between repeats lead to the breakdown of co-operativity and the appearance of folding sub-domains within an apparently regular repeat array. The quasi-1D architecture of repeat-proteins is crucial in detailing how the local energetic balances can modulate the folding dynamics of these proteins, which can be related to the physiological behaviour of these ubiquitous biological systems. Introduction and between repeats, challenging the concept of structural It was early on noted that many natural proteins typically domain. collapse stretches of amino acid chains into compact units, defining structural domains [1]. These domains typically correlate with biological activities and many modern proteins can be described as composed by novel ‘domain arrange- Definition of the repeat-units ments’ [2]. -

A Structural Guide to Proteins of the NF-Kb Signaling Module
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A Structural Guide to Proteins of the NF-kB Signaling Module Tom Huxford1 and Gourisankar Ghosh2 1Department of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182-1030 2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0375 Correspondence: [email protected] The prosurvival transcription factor NF-kB specifically binds promoter DNA to activate target gene expression. NF-kB is regulated through interactions with IkB inhibitor proteins. Active proteolysis of these IkB proteins is, in turn, under the control of the IkB kinase complex (IKK). Together, these three molecules form the NF-kB signaling module. Studies aimed at charac- terizing the molecular mechanisms of NF-kB, IkB, and IKK in terms of their three-dimen- sional structures have lead to a greater understanding of this vital transcription factor system. F-kB is a master transcription factor that from the perspective of their three-dimensional Nresponds to diverse cell stimuli by activat- structures. ing the expression of stress response genes. Multiple signals, including cytokines, growth factors, engagement of the T-cell receptor, and NF-kB bacterial and viral products, induce NF-kB Introduction to NF-kB transcriptional activity (Hayden and Ghosh 2008). A point of convergence for the myriad NF-kB was discovered in the laboratory of of NF-kB inducing signals is the IkB kinase David Baltimore as a nuclear activity with bind- complex (IKK). Active IKK in turn controls ing specificity toward a ten-base-pair DNA transcription factor NF-kB by regulating pro- sequence 50-GGGACTTTCC-30 present within teolysis of the IkB inhibitor protein (Fig. -
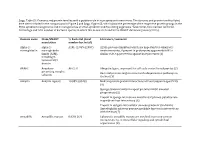
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Using Small Globular Proteins to Study Folding Stability and Aggregation
Using Small Globular Proteins to Study Folding Stability and Aggregation Virginia Castillo Cano September 2012 Universitat Autònoma de Barcelona Departament de Bioquímica i Biologia Molecular Institut de Biotecnologia i de Biomedicina Using Small Globular Proteins to Study Folding Stability and Aggregation Doctoral thesis presented by Virginia Castillo Cano for the degree of PhD in Biochemistry, Molecular Biology and Biomedicine from the University Autonomous of Barcelona. Thesis performed in the Department of Biochemistry and Molecular Biology, and Institute of Biotechnology and Biomedicine, supervised by Dr. Salvador Ventura Zamora. Virginia Castillo Cano Salvador Ventura aZamor Cerdanyola del Vallès, September 2012 SUMMARY SUMMARY The purpose of the thesis entitled “Using small globular proteins to study folding stability and aggregation” is to contribute to understand how globular proteins fold into their native, functional structures or, alternatively, misfold and aggregate into toxic assemblies. Protein misfolding diseases include an important number of human disorders such as Parkinson’s and Alzheimer’s disease, which are related to conformational changes from soluble non‐toxic to aggregated toxic species. Moreover, the over‐expression of recombinant proteins usually leads to the accumulation of protein aggregates, being a major bottleneck in several biotechnological processes. Hence, the development of strategies to diminish or avoid these taberran reactions has become an important issue in both biomedical and biotechnological industries. In the present thesis we have used a battery of biophysical and computational techniques to analyze the folding, conformational stability and aggregation propensity of several globular proteins. The combination of experimental (in vivo and in vitro) and bioinformatic approaches has provided insights into the intrinsic and structural properties, including the presence of disulfide bonds and the quaternary structure, that modulate these processes under physiological conditions. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Armc5 Deletion Causes Developmental Defects and Compromises T-Cell Immune Responses
ARTICLE Received 26 Jan 2016 | Accepted 4 Nov 2016 | Published 7 Feb 2017 DOI: 10.1038/ncomms13834 OPEN Armc5 deletion causes developmental defects and compromises T-cell immune responses Yan Hu 1,*, Linjiang Lao1,*, Jianning Mao1, Wei Jin1, Hongyu Luo1, Tania Charpentier2, Shijie Qi1, Junzheng Peng1, Bing Hu3, Mieczyslaw Martin Marcinkiewicz4, Alain Lamarre2 & Jiangping Wu1,5 Armadillo repeat containing 5 (ARMC5) is a cytosolic protein with no enzymatic activities. Little is known about its function and mechanisms of action, except that gene mutations are associated with risks of primary macronodular adrenal gland hyperplasia. Here we map Armc5 expression by in situ hybridization, and generate Armc5 knockout mice, which are small in body size. Armc5 knockout mice have compromised T-cell proliferation and differentiation into Th1 and Th17 cells, increased T-cell apoptosis, reduced severity of experimental autoimmune encephalitis, and defective immune responses to lymphocytic choriomeningitis virus infection. These mice also develop adrenal gland hyperplasia in old age. Yeast 2-hybrid assays identify 16 ARMC5-binding partners. Together these data indicate that ARMC5 is crucial in fetal development, T-cell function and adrenal gland growth homeostasis, and that the functions of ARMC5 probably depend on interaction with multiple signalling pathways. 1 Centre de recherche (CR), Centre hospitalier de l’Universite´ de Montre´al (CHUM), 900 Rue Saint Denis, Montre´al, Que´bec, Canada H2X 0A9. 2 Institut national de la recherche scientifique—Institut Armand-Frappier (INRS-IAF), 531 Boul. des Prairies, Laval, Que´bec, Canada H7V 1B7. 3 Anatomic Pathology, AmeriPath Central Florida, 4225 Fowler Ave. Tampa, Orlando, Florida 33617, USA. -

WSB1: from Homeostasis to Hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew Macisaac1,2,3 and Douglas James Demetrick1,2,3,4*
Haque et al. Journal of Biomedical Science (2016) 23:61 DOI 10.1186/s12929-016-0270-3 REVIEW Open Access WSB1: from homeostasis to hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew MacIsaac1,2,3 and Douglas James Demetrick1,2,3,4* Abstract The wsb1 gene has been identified to be important in developmental biology and cancer. A complex transcriptional regulation of wsb1 yields at least three functional transcripts. The major expressed isoform, WSB1 protein, is a substrate recognition protein within an E3 ubiquitin ligase, with the capability to bind diverse targets and mediate ubiquitinylation and proteolytic degradation. Recent data suggests a new role for WSB1 as a component of a neuroprotective pathway which results in modification and aggregation of neurotoxic proteins such as LRRK2 in Parkinson’s Disease, via an unusual mode of protein ubiquitinylation. WSB1 is also involved in thyroid hormone homeostasis, immune regulation and cellular metabolism, particularly glucose metabolism and hypoxia. In hypoxia, wsb1 is a HIF-1 target, and is a regulator of the degradation of diverse proteins associated with the cellular response to hypoxia, including HIPK2, RhoGDI2 and VHL. Major roles are to both protect HIF-1 function through degradation of VHL, and decrease apoptosis through degradation of HIPK2. These activities suggest a role for wsb1 in cancer cell proliferation and metastasis. As well, recent work has identified a role for WSB1 in glucose metabolism, and perhaps in mediating the Warburg effect in cancer cells by maintaining the function of HIF1. Furthermore, studies of cancer specimens have identified dysregulation of wsb1 associated with several types of cancer, suggesting a biologically relevant role in cancer development and/or progression. -

Topology Is the Principal Determinant in the Folding of a Complex All-Alpha Greek Key Death Domain from Human FADD
doi:10.1016/j.jmb.2009.04.004 J. Mol. Biol. (2009) 389, 425–437 Available online at www.sciencedirect.com Topology is the Principal Determinant in the Folding of a Complex All-alpha Greek Key Death Domain from Human FADD Annette Steward, Gary S. McDowell and Jane Clarke⁎ University of Cambridge In order to elucidate the relative importance of secondary structure and Department of Chemistry, MRC topology in determining folding mechanism, we have carried out a phi- Centre for Protein Engineering, value analysis of the death domain (DD) from human FADD. FADD DD is a Lensfield Road, Cambridge, CB2 100 amino acid domain consisting of six anti-parallel alpha helices arranged 1EW, UK in a Greek key structure. We asked how does the folding of this domain compare with that of (a) other all-alpha-helical proteins and (b) other Greek Received 11 February 2009; key proteins? Is the folding pathway determined mainly by secondary received in revised form structure or is topology the principal determinant? Our Φ-value analysis 26 March 2009; reveals a striking resemblance to the all-beta Greek key immunoglobulin- accepted 1 April 2009 like domains. Both fold via diffuse transition states and, importantly, long- Available online range interactions between the four central elements of secondary structure 9 April 2009 are established in the transition state. The elements of secondary structure that are less tightly associated with the central core are less well packed in both cases. Topology appears to be the dominant factor in determining the pathway of folding in all Greek key domains. -

SAP Domain Phi-Value Analysis
Protein folding of the SAP domain, a naturally occurring two-helix bundle Charlotte A Dodson 1,2 & Eyal Arbely 1,3 1MRC Centre for Protein Engineering, Hills Road, Cambridge CB2 0QH, UK 2Current address: Molecular Medicine, National Heart and Lung Institute, Imperial College London, SAF Building, London SW7 2AZ, UK 3Current address: Department of Chemistry and National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel Corresponding author: Charlotte A Dodson, Molecular Medicine, National Heart & Lung Institute, Imperial College London, SAF Building, London SW7 2AZ, UK; e-mail: [email protected]; tel: +44 20 7594 3053. The SAP domain from the Saccharomyces cerevisiae Tho1 protein is comprised of just two helices and a hydrophobic core and is one of the smallest proteins whose folding has been characterised. Φ-value analysis revealed that Tho1 SAP folds through a transition state where helix 1 is the most extensively formed element of secondary structure and flickering native-like core contacts from Leu35 are also present. The contacts that contribute most to native state stability of Tho1 SAP are not formed in the transition state. Keywords: SAP domain, protein folding, Φ-value, transition state analysis, Tho1 1 Highlights • Tho1 SAP domain is one of the smallest model protein folding domains • SAP domain folds through a diffuse transition state in which helix 1 is most formed • Native state stability is dominated by contacts formed after the transition state 2 Introduction The principles which govern the folding and unfolding of proteins have fascinated the scientific community for decades [1]. -

The Effect of Additional Disulfide Bonds on the Stability and Folding of Ribonuclease a Pascal Pecher, Ulrich Arnold
The effect of additional disulfide bonds on the stability and folding of ribonuclease A Pascal Pecher, Ulrich Arnold To cite this version: Pascal Pecher, Ulrich Arnold. The effect of additional disulfide bonds on the stability and folding of ribonuclease A. Biophysical Chemistry, Elsevier, 2009, 141 (1), pp.21. 10.1016/j.bpc.2008.12.005. hal-00531136 HAL Id: hal-00531136 https://hal.archives-ouvertes.fr/hal-00531136 Submitted on 2 Nov 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ÔØ ÅÒÙ×Ö ÔØ The effect of additional disulfide bonds on the stability and folding of ribonuclease A Pascal Pecher, Ulrich Arnold PII: S0301-4622(08)00273-1 DOI: doi: 10.1016/j.bpc.2008.12.005 Reference: BIOCHE 5203 To appear in: Biophysical Chemistry Received date: 29 September 2008 Revised date: 11 December 2008 Accepted date: 13 December 2008 Please cite this article as: Pascal Pecher, Ulrich Arnold, The effect of additional disulfide bonds on the stability and folding of ribonuclease A, Biophysical Chemistry (2009), doi: 10.1016/j.bpc.2008.12.005 This is a PDF file of an unedited manuscript that has been accepted for publication.