Dynamics in Folded and Unfolded Peptides and Proteins Measured by Triplet-Triplet Energy Transfer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanism of Resilin Elasticity
ARTICLE Received 24 Oct 2011 | Accepted 11 Jul 2012 | Published 14 Aug 2012 DOI: 10.1038/ncomms2004 Mechanism of resilin elasticity Guokui Qin1,*, Xiao Hu1,*, Peggy Cebe2 & David L. Kaplan1 Resilin is critical in the flight and jumping systems of insects as a polymeric rubber-like protein with outstanding elasticity. However, insight into the underlying molecular mechanisms responsible for resilin elasticity remains undefined. Here we report the structure and function of resilin from Drosophila CG15920. A reversible beta-turn transition was identified in the peptide encoded by exon III and for full-length resilin during energy input and release, features that correlate to the rapid deformation of resilin during functions in vivo. Micellar structures and nanoporous patterns formed after beta-turn structures were present via changes in either the thermal or the mechanical inputs. A model is proposed to explain the super elasticity and energy conversion mechanisms of resilin, providing important insight into structure–function relationships for this protein. Furthermore, this model offers a view of elastomeric proteins in general where beta-turn-related structures serve as fundamental units of the structure and elasticity. 1 Department of Biomedical Engineering, Tufts University, Medford, Massachusetts 02155, USA. 2 Department of Physics and Astronomy, Tufts University, Medford, Massachusetts 02155, USA. *These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.L.K. (email: [email protected]). -
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
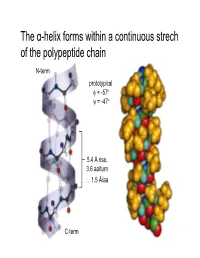
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Using Small Globular Proteins to Study Folding Stability and Aggregation
Using Small Globular Proteins to Study Folding Stability and Aggregation Virginia Castillo Cano September 2012 Universitat Autònoma de Barcelona Departament de Bioquímica i Biologia Molecular Institut de Biotecnologia i de Biomedicina Using Small Globular Proteins to Study Folding Stability and Aggregation Doctoral thesis presented by Virginia Castillo Cano for the degree of PhD in Biochemistry, Molecular Biology and Biomedicine from the University Autonomous of Barcelona. Thesis performed in the Department of Biochemistry and Molecular Biology, and Institute of Biotechnology and Biomedicine, supervised by Dr. Salvador Ventura Zamora. Virginia Castillo Cano Salvador Ventura aZamor Cerdanyola del Vallès, September 2012 SUMMARY SUMMARY The purpose of the thesis entitled “Using small globular proteins to study folding stability and aggregation” is to contribute to understand how globular proteins fold into their native, functional structures or, alternatively, misfold and aggregate into toxic assemblies. Protein misfolding diseases include an important number of human disorders such as Parkinson’s and Alzheimer’s disease, which are related to conformational changes from soluble non‐toxic to aggregated toxic species. Moreover, the over‐expression of recombinant proteins usually leads to the accumulation of protein aggregates, being a major bottleneck in several biotechnological processes. Hence, the development of strategies to diminish or avoid these taberran reactions has become an important issue in both biomedical and biotechnological industries. In the present thesis we have used a battery of biophysical and computational techniques to analyze the folding, conformational stability and aggregation propensity of several globular proteins. The combination of experimental (in vivo and in vitro) and bioinformatic approaches has provided insights into the intrinsic and structural properties, including the presence of disulfide bonds and the quaternary structure, that modulate these processes under physiological conditions. -

Topology Is the Principal Determinant in the Folding of a Complex All-Alpha Greek Key Death Domain from Human FADD
doi:10.1016/j.jmb.2009.04.004 J. Mol. Biol. (2009) 389, 425–437 Available online at www.sciencedirect.com Topology is the Principal Determinant in the Folding of a Complex All-alpha Greek Key Death Domain from Human FADD Annette Steward, Gary S. McDowell and Jane Clarke⁎ University of Cambridge In order to elucidate the relative importance of secondary structure and Department of Chemistry, MRC topology in determining folding mechanism, we have carried out a phi- Centre for Protein Engineering, value analysis of the death domain (DD) from human FADD. FADD DD is a Lensfield Road, Cambridge, CB2 100 amino acid domain consisting of six anti-parallel alpha helices arranged 1EW, UK in a Greek key structure. We asked how does the folding of this domain compare with that of (a) other all-alpha-helical proteins and (b) other Greek Received 11 February 2009; key proteins? Is the folding pathway determined mainly by secondary received in revised form structure or is topology the principal determinant? Our Φ-value analysis 26 March 2009; reveals a striking resemblance to the all-beta Greek key immunoglobulin- accepted 1 April 2009 like domains. Both fold via diffuse transition states and, importantly, long- Available online range interactions between the four central elements of secondary structure 9 April 2009 are established in the transition state. The elements of secondary structure that are less tightly associated with the central core are less well packed in both cases. Topology appears to be the dominant factor in determining the pathway of folding in all Greek key domains. -

SAP Domain Phi-Value Analysis
Protein folding of the SAP domain, a naturally occurring two-helix bundle Charlotte A Dodson 1,2 & Eyal Arbely 1,3 1MRC Centre for Protein Engineering, Hills Road, Cambridge CB2 0QH, UK 2Current address: Molecular Medicine, National Heart and Lung Institute, Imperial College London, SAF Building, London SW7 2AZ, UK 3Current address: Department of Chemistry and National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel Corresponding author: Charlotte A Dodson, Molecular Medicine, National Heart & Lung Institute, Imperial College London, SAF Building, London SW7 2AZ, UK; e-mail: [email protected]; tel: +44 20 7594 3053. The SAP domain from the Saccharomyces cerevisiae Tho1 protein is comprised of just two helices and a hydrophobic core and is one of the smallest proteins whose folding has been characterised. Φ-value analysis revealed that Tho1 SAP folds through a transition state where helix 1 is the most extensively formed element of secondary structure and flickering native-like core contacts from Leu35 are also present. The contacts that contribute most to native state stability of Tho1 SAP are not formed in the transition state. Keywords: SAP domain, protein folding, Φ-value, transition state analysis, Tho1 1 Highlights • Tho1 SAP domain is one of the smallest model protein folding domains • SAP domain folds through a diffuse transition state in which helix 1 is most formed • Native state stability is dominated by contacts formed after the transition state 2 Introduction The principles which govern the folding and unfolding of proteins have fascinated the scientific community for decades [1]. -

Synthesis and Nmr Studies of a Β-Turn Mimetic Molecular
SYNTHESIS AND NMR STUDIES OF A β-TURN MIMETIC MOLECULAR TORSION BALANCE by Melissa Ann Liberatore B.S. Chemistry, Lehigh University, 2006 Submitted to the Graduate Faculty of the Kenneth P. Dietrich School of Arts and Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2012 UNIVERSITY OF PITTSBURGH DIETRICH SCHOOL OF ARTS AND SCIENCES This dissertation was presented by Melissa Ann Liberatore It was defended on July 23, 2012 and approved by Professor Dennis Curran, Department of Chemistry Professor Michael Trakselis, Department of Chemistry Professor Judith Klein-Seetharaman, Department of Structural Biology Dissertation Advisor: Professor Craig Wilcox, Department of Chemistry ii Copyright © by Melissa Ann Liberatore 2012 iii SYNTHESIS AND NMR STUDIES OF A β-TURN MIMETIC MOLECULAR TORSION BALANCE Melissa Ann Liberatore, PhD University of Pittsburgh, 2012 The attainment of precise measurements of the molecular forces that influence protein folding is important in order to further understand peptide dynamics and stability. A hybrid synthetic- natural peptide motif, combining an o,o,o’-trisubstituted biphenyl with an (ortho-tolyl)-amide, was synthesized in multiple formats and studied by NMR to probe the effects of amino acid substitutions on antiparallel beta-sheet configuration and stability. The potential of this “molecular torsion balance” as a beta-turn mimic was demonstrated by quantifying the rotational barriers about several axes. The free-energy rotational barrier of the aryl-aryl bond was found to be 35.7 kcal mol-1 at 418 K in hexanes. EXSY analysis was also used to measure barriers about -1 -1 the N-aryl (20.9 kcal mol at 343 K in toluene-d8) and N-CO bonds (17.2 kcal mol at 298 K in chloroform-d). -

Duke University Dissertation Template
Order and Disorder in Protein Biomaterial Design by Stefan Daniel Roberts Department of Biomedical Engineering Duke University Date:_______________________ Approved: ___________________________ Ashutosh Chilkoti, Supervisor ___________________________ Joel Collier ___________________________ Brenton Hoffman ___________________________ Terrence Oas ___________________________ Rohit Pappu Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biomedical Engineering in the Graduate School of Duke University 2018 ABSTRACT Order and Disorder in Protein Biomaterial Design by Stefan Daniel Roberts Department of Biomedical Engineering Duke University Date:_______________________ Approved: ___________________________ Ashutosh Chilkoti, Supervisor ___________________________ Joel Collier ___________________________ Brenton Hoffman ___________________________ Terrence Oas ___________________________ Rohit Pappu An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biomedical Engineering in the Graduate School of Duke University 2018 Copyright by Stefan Daniel Roberts 2018 Abstract Crystalline and amorphous materials have been extensively studied for their interesting properties, but they comprise a very small portion of the total materials space. The properties of most materials are a consequence of the interactions between their ordered and disordered components. This phenomenon is especially -

The Effect of Additional Disulfide Bonds on the Stability and Folding of Ribonuclease a Pascal Pecher, Ulrich Arnold
The effect of additional disulfide bonds on the stability and folding of ribonuclease A Pascal Pecher, Ulrich Arnold To cite this version: Pascal Pecher, Ulrich Arnold. The effect of additional disulfide bonds on the stability and folding of ribonuclease A. Biophysical Chemistry, Elsevier, 2009, 141 (1), pp.21. 10.1016/j.bpc.2008.12.005. hal-00531136 HAL Id: hal-00531136 https://hal.archives-ouvertes.fr/hal-00531136 Submitted on 2 Nov 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ÔØ ÅÒÙ×Ö ÔØ The effect of additional disulfide bonds on the stability and folding of ribonuclease A Pascal Pecher, Ulrich Arnold PII: S0301-4622(08)00273-1 DOI: doi: 10.1016/j.bpc.2008.12.005 Reference: BIOCHE 5203 To appear in: Biophysical Chemistry Received date: 29 September 2008 Revised date: 11 December 2008 Accepted date: 13 December 2008 Please cite this article as: Pascal Pecher, Ulrich Arnold, The effect of additional disulfide bonds on the stability and folding of ribonuclease A, Biophysical Chemistry (2009), doi: 10.1016/j.bpc.2008.12.005 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Different Members of a Simple Three-Helix Bundle Protein Family Have Very Different Folding Rate Constants and Fold by Different Mechanisms
doi:10.1016/j.jmb.2009.05.010 J. Mol. Biol. (2009) 390, 1074–1085 Available online at www.sciencedirect.com Different Members of a Simple Three-Helix Bundle Protein Family Have Very Different Folding Rate Constants and Fold by Different Mechanisms Beth G. Wensley, Martina Gärtner, Wan Xian Choo, Sarah Batey and Jane Clarke⁎ Department of Chemistry, MRC The 15th, 16th, and 17th repeats of chicken brain α-spectrin (R15, R16, and Centre for Protein Engineering, R17, respectively) are very similar in terms of structure and stability. University of Cambridge, However, R15 folds and unfolds 3 orders of magnitude faster than R16 and Lensfield Road, Cambridge CB2 R17. This is unexpected. The rate-limiting transition state for R15 folding is 1EW, UK investigated using protein engineering methods (Φ-value analysis) and compared with previously completed analyses of R16 and R17. Character- Received 10 February 2009; isation of many mutants suggests that all three proteins have similar received in revised form complexity in the folding landscape. The early rate-limiting transition states 5 May 2009; of the three domains are similar in terms of overall structure, but there are accepted 8 May 2009 significant differences in the patterns of Φ-values. R15 apparently folds via a Available online nucleation–condensation mechanism, which involves concomitant folding 13 May 2009 and packing of the A- and C-helices, establishing the correct topology. R16 and R17 fold via a more framework-like mechanism, which may impede the search to find the correct packing of the helices, providing a possible explanation for the fast folding of R15. -

Folding Versus Aggregation: Polypeptide Conformations on Competing Pathways
Available online at www.sciencedirect.com ABB Archives of Biochemistry and Biophysics 469 (2008) 100–117 www.elsevier.com/locate/yabbi Review Folding versus aggregation: Polypeptide conformations on competing pathways Thomas R. Jahn, Sheena E. Radford * Astbury Centre for Structural and Molecular Biology, Institute of Molecular and Cellular Biology, University of Leeds, Mount Preston Street, Leeds LS2 9JT, UK Received 3 April 2007, and in revised form 16 May 2007 Available online 8 June 2007 Abstract Protein aggregation has now become recognised as an important and generic aspect of protein energy landscapes. Since the discovery that numerous human diseases are caused by protein aggregation, the biophysical characterisation of misfolded states and their aggre- gation mechanisms has received increased attention. Utilising experimental techniques and computational approaches established for the analysis of protein folding reactions has ensured rapid advances in the study of pathways leading to amyloid fibrils and amyloid-related aggregates. Here we describe recent experimental and theoretical advances in the elucidation of the conformational properties of dynamic, heterogeneous and/or insoluble protein ensembles populated on complex, multidimensional protein energy landscapes. We dis- cuss current understanding of aggregation mechanisms in this context and describe how the synergy between biochemical, biophysical and cell-biological experiments are beginning to provide detailed insights into the partitioning of non-native species between -

Identification and Characterization of Protein Folding Intermediates Stefano Gianni, Ylva Ivarsson, Per Jemth, Maurizio Brunori, Carlo Travaglini-Allocatelli
Identification and characterization of protein folding intermediates Stefano Gianni, Ylva Ivarsson, Per Jemth, Maurizio Brunori, Carlo Travaglini-Allocatelli To cite this version: Stefano Gianni, Ylva Ivarsson, Per Jemth, Maurizio Brunori, Carlo Travaglini-Allocatelli. Identifica- tion and characterization of protein folding intermediates. Biophysical Chemistry, Elsevier, 2007, 128 (2-3), pp.105. 10.1016/j.bpc.2007.04.008. hal-00501662 HAL Id: hal-00501662 https://hal.archives-ouvertes.fr/hal-00501662 Submitted on 12 Jul 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ÔØ ÅÒÙ×Ö ÔØ Identification and characterization of protein folding intermediates Stefano Gianni, Ylva Ivarsson, Per Jemth, Maurizio Brunori, Carlo Travaglini- Allocatelli PII: S0301-4622(07)00087-7 DOI: doi: 10.1016/j.bpc.2007.04.008 Reference: BIOCHE 4952 To appear in: Biophysical Chemistry Received date: 29 March 2007 Revised date: 16 April 2007 Accepted date: 16 April 2007 Please cite this article as: Stefano Gianni, Ylva Ivarsson, Per Jemth, Maurizio Brunori, Carlo Travaglini-Allocatelli, Identification and characterization of protein folding inter- mediates, Biophysical Chemistry (2007), doi: 10.1016/j.bpc.2007.04.008 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Fibrillar Self-Assembly of a Chimeric Elastin-Resilin Inspired Engineered Polypeptide
nanomaterials Article Fibrillar Self-Assembly of a Chimeric Elastin-Resilin Inspired Engineered Polypeptide Angelo Bracalello 1, Valeria Secchi 2,* , Roberta Mastrantonio 2, Antonietta Pepe 1, Tiziana Persichini 2, Giovanna Iucci 2 , Brigida Bochicchio 1,* and Chiara Battocchio 2,* 1 Department of Sciences, University of Basilicata, Via Ateneo Lucano, 10, 85100 Potenza, Italy; [email protected] (A.B.); [email protected] (A.P.) 2 Department of Sciences, University of Roma Tre, Via della Vasca Navale, 79, 00146 Rome, Italy; [email protected] (R.M.); [email protected] (T.P.); [email protected] (G.I.) * Correspondence: [email protected] (V.S.); [email protected] (B.B.); [email protected] (C.B.); Tel.: +39-06-5733-3400 (C.B.) Received: 2 October 2019; Accepted: 11 November 2019; Published: 14 November 2019 Abstract: In the field of tissue engineering, recombinant protein-based biomaterials made up of block polypeptides with tunable properties arising from the functionalities of the individual domains are appealing candidates for the construction of medical devices. In this work, we focused our attention on the preparation and structural characterization of nanofibers from a chimeric-polypeptide-containing resilin and elastin domain, designed on purpose to enhance its cell-binding ability by introducing a specific fibronectin-derived Arg-Gly-Asp (RGD) sequence. The polypeptide ability to self-assemble was investigated. The molecular and supramolecular structure was characterized by Scanning Electronic Microscopy (SEM) and Atomic Force Microscopy (AFM), circular dichroism, state-of-the-art synchrotron radiation-induced techniques X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure spectroscopy (NEXAFS).