The Structure of Small Beta Barrels
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Simulations of Я-Hairpin Folding Confined to Spherical Pores Using
Simulations of -hairpin folding confined to spherical pores using distributed computing D. K. Klimov*†, D. Newfield‡, and D. Thirumalai*† *Institute for Physical Science and Technology, University of Maryland, College Park, MD 20742; and ‡Parabon Computation, 3930 Walnut Street, Suite 100, Fairfax, VA 22030-4738 Communicated by George H. Lorimer, University of Maryland, College Park, MD, April 12, 2002 (received for review December 18, 2001) 3 ϱ We report the thermodynamics and kinetics of an off-lattice Go radius of gyration of a chain Rg at D (the size of a chain in  Ӎ model -hairpin from Ig-binding protein confined to an inert bulk solution) to N according to Rg aN . If the chain is ideal, ϭ ⌬ ϭ ͞ 2 spherical pore. Confinement enhances the stability of the hairpin then 0.5 and FU RTN(a D) . Because of the reduction due to the decrease in the entropy of the unfolded state. Compared in the translational entropy, confinement also increases the free ⌬ Ͼ ⌬ ͞⌬ ϽϽ with their values in the bulk, the rates of hairpin formation increase energy of the native state, i.e., FN 0. If FN FU 1, then in the spherical pore. Surprisingly, the dependence of the rates on localization of a protein in a confined space stabilizes the native the pore radius, Rs, is nonmonotonic. The rates reach a maximum state compared with the bulk. It also follows that there is a range ͞ b Ӎ b at Rs Rg,N 1.5, where Rg,N is the radius of gyration of the folded of D values over which stability is maximized. -

Evolution of Biomolecular Structure Class II Trna-Synthetases and Trna
University of Illinois at Urbana-Champaign Luthey-Schulten Group Theoretical and Computational Biophysics Group Evolution of Biomolecular Structure Class II tRNA-Synthetases and tRNA MultiSeq Developers: Prof. Zan Luthey-Schulten Elijah Roberts Patrick O’Donoghue John Eargle Anurag Sethi Dan Wright Brijeet Dhaliwal September 25, 2006. A current version of this tutorial is available at http://www.scs.uiuc.edu/˜schulten/tutorials/evolution/ CONTENTS 2 Contents 1 Introduction 4 1.1 The MultiSeq Bioinformatic Analysis Environment . 4 1.2 Aminoacyl-tRNA Synthetases: Role in translation . 4 1.3 Getting Started . 7 1.3.1 Requirements . 7 1.3.2 Copying the tutorial files . 7 1.3.3 Configuring MultiSeq . 7 1.3.4 Configuring BLAST for MultiSeq . 10 1.4 The Aspartyl-tRNA Synthetase/tRNA Complex . 12 1.4.1 Loading the structure into MultiSeq . 12 1.4.2 Selecting and highlighting residues . 13 1.4.3 Domain organization of the synthetase . 14 1.4.4 Nearest neighbor contacts . 14 2 Evolutionary Analysis of AARS Structures 17 2.1 Loading Molecules . 17 2.2 Multiple Structure Alignments . 18 2.3 Structural Conservation Measure: Qres . 19 2.4 Structure Based Phylogenetic Analysis . 21 2.4.1 Limitations of sequence data . 21 2.4.2 Structural metrics look further back in time . 23 3 Complete Evolutionary Profile of AspRS 26 3.1 BLASTing Sequence Databases . 26 3.1.1 Importing the archaeal sequences . 26 3.1.2 Now the other domains of life . 27 3.2 Organizing Your Data . 28 3.3 Finding a Structural Domain in a Sequence . 29 3.4 Aligning to a Structural Profile using ClustalW . -

The Architecture of the Protein Domain Universe
The architecture of the protein domain universe Nikolay V. Dokholyan Department of Biochemistry and Biophysics, The University of North Carolina at Chapel Hill, School of Medicine, Chapel Hill, NC 27599 ABSTRACT Understanding the design of the universe of protein structures may provide insights into protein evolution. We study the architecture of the protein domain universe, which has been found to poses peculiar scale-free properties (Dokholyan et al., Proc. Natl. Acad. Sci. USA 99: 14132-14136 (2002)). We examine the origin of these scale-free properties of the graph of protein domain structures (PDUG) and determine that that the PDUG is not modular, i.e. it does not consist of modules with uniform properties. Instead, we find the PDUG to be self-similar at all scales. We further characterize the PDUG architecture by studying the properties of the hub nodes that are responsible for the scale-free connectivity of the PDUG. We introduce a measure of the betweenness centrality of protein domains in the PDUG and find a power-law distribution of the betweenness centrality values. The scale-free distribution of hubs in the protein universe suggests that a set of specific statistical mechanics models, such as the self-organized criticality model, can potentially identify the principal driving forces of molecular evolution. We also find a gatekeeper protein domain, removal of which partitions the largest cluster into two large sub- clusters. We suggest that the loss of such gatekeeper protein domains in the course of evolution is responsible for the creation of new fold families. INTRODUCTION The principles of molecular evolution remain elusive despite fundamental breakthroughs on the theoretical front 1-5 and a growing amount of genomic and proteomic data, over 23,000 solved protein structures 6 and protein functional annotations 7-9. -

The TIM Barrel Fold Nagarajan D
The TIM barrel fold Nagarajan D. and Nanajkar N. Comments and corrections: Line 10: fix “αhelices” in “α-helices”. Lines 11-12: C-terminal loops are important for catalytic activity, while N-terminal loops are important for the stability of the TIM-barrels. This should be mentioned. Line 14: The reference #7 is not related to the statement. Line 14: There is a new EC classe (EC.7, translocases). Change “5 of 6” in “5 of 7”. Lines 26-27: It is not correct to state that the shear number of 8 for the TIM-barrels is due to “their staggered nature”. Most of the β-barrels have a staggered nature, but their shear number is not 8. Line 27: The reference #2 is imprecise. Wierenga did not defined himself the shear number of TIM-barrel proteins. Please check the 2 papers of Murzin AG, 1994, “Principle determining the structure of β-sheet barrels in proteins,” I and II, and the paper of Liu W, 1998, “Shear numbers of protein β-barrels: definition refinements and statistics”. Line 29: Again, it is not correct to state that the 4-fold geometric symmetry depends on the stagger. Since the number of strands (n) is equal to the Shear number (S), side-chains point alternatively towards the pore and the core, giving a 4-fold symmetry. Line 37: “historically” is a bit exaggerated for a reference dated 2015, especially if it comes from the author itself. Find a true historic reference, or just mention that you defined the regions “core” and “pore”. Line 43: “Consequently” is misleading. -

Smurflite: Combining Simplified Markov Random Fields With
SMURFLite: combining simplified Markov random fields with simulated evolution improves remote homology detection for beta-structural proteins into the twilight zone Noah M. Daniels 1, Raghavendra Hosur 2, Bonnie Berger 2∗, and Lenore J. Cowen 1∗ 1Department of Computer Science, Tufts University, Medford, MA 02155 2Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA 02139 ABSTRACT are limited in their power to recognize remote homologs because of Motivation: One of the most successful methods to date for their inability to model statistical dependencies between amino-acid recognizing protein sequences that are evolutionarily related has residues that are close in space but far apart in sequence (Lifson and been profile Hidden Markov Models (HMMs). However, these models Sander (1980); Zhu and Braun (1999); Olmea et al. (1999); Cowen do not capture pairwise statistical preferences of residues that are et al. (2002); Steward and Thorton (2002)). hydrogen bonded in beta sheets. These dependencies have been For this reason, many have suggested (White et al. (1994); partially captured in the HMM setting by simulated evolution in the Lathrop and Smith (1996); Thomas et al. (2008); Liu et al. (2009); training phase and can be fully captured by Markov Random Fields Menke et al. (2010); Peng and Xu (2011)) that more powerful (MRFs). However, the MRFs can be computationally prohibitive when Markov Random Fields (MRFs) be used. MRFs employ an auxiliary beta strands are interleaved in complex topologies. dependency graph which allows them to model more complex We introduce SMURFLite, a method that combines both simplified statistical dependencies, including statistical dependencies that Markov Random Fields and simulated evolution to substantially occur between amino-acid residues that are hydrogen bonded in beta improve remote homology detection for beta structures. -

Documentation and Localization of Force-Mediated Filamin a Domain
ARTICLE Received 29 May 2014 | Accepted 10 Jul 2014 | Published 14 Aug 2014 DOI: 10.1038/ncomms5656 Documentation and localization of force-mediated filamin A domain perturbations in moving cells Fumihiko Nakamura1, Mia Song1, John H. Hartwig1 & Thomas P. Stossel1 Endogenously and externally generated mechanical forces influence diverse cellular activities, a phenomenon defined as mechanotransduction. Deformation of protein domains by application of stress, previously documented to alter macromolecular interactions in vitro, could mediate these effects. We engineered a photon-emitting system responsive to unfolding of two repeat domains of the actin filament (F-actin) crosslinker protein filamin A (FLNA) that binds multiple partners involved in cell signalling reactions and validated the system using F-actin networks subjected to myosin-based contraction. Expressed in cultured cells, the sensor-containing FLNA construct reproducibly reported FLNA domain unfolding strikingly localized to dynamic, actively protruding, leading cell edges. The unfolding signal depends upon coherence of F-actin-FLNA networks and is enhanced by stimulating cell contractility. The results establish protein domain distortion as a bona fide mechanism for mechanotransduction in vivo. 1 Translational Medicine Division, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02445, USA. Correspondence and requests for materials should be addressed to F.N. (email: [email protected]). NATURE COMMUNICATIONS | 5:4656 | DOI: 10.1038/ncomms5656 -

Lipid-Targeting Pleckstrin Homology Domain Turns Its Autoinhibitory Face Toward the TEC Kinases
Lipid-targeting pleckstrin homology domain turns its autoinhibitory face toward the TEC kinases Neha Amatyaa, Thomas E. Walesb, Annie Kwonc, Wayland Yeungc, Raji E. Josepha, D. Bruce Fultona, Natarajan Kannanc, John R. Engenb, and Amy H. Andreottia,1 aRoy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, IA 50011; bDepartment of Chemistry and Chemical Biology, Northeastern University, Boston, MA 02115; and cInstitute of Bioinformatics and Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602 Edited by Natalie G. Ahn, University of Colorado Boulder, Boulder, CO, and approved September 17, 2019 (received for review May 3, 2019) The pleckstrin homology (PH) domain is well known for its phos- activation loop phosphorylation site are also controlled by noncatalytic pholipid targeting function. The PH-TEC homology (PHTH) domain domains (17). In addition to the N-terminal PHTH domain, the within the TEC family of tyrosine kinases is also a crucial component TEC kinases contain a proline-rich region (PRR) and Src ho- of the autoinhibitory apparatus. The autoinhibitory surface on the mology 3 (SH3) and Src homology 2 (SH2) domains that impinge PHTH domain has been previously defined, and biochemical investi- on the kinase domain to alter the conformational ensemble and gations have shown that PHTH-mediated inhibition is mutually thus the activation status of the enzyme. A crystal structure of the exclusive with phosphatidylinositol binding. Here we use hydrogen/ BTK SH3-SH2-kinase fragment has been solved (10) showing that deuterium exchange mass spectrometry, nuclear magnetic resonance the SH3 and SH2 domains of BTK assemble onto the distal side of (NMR), and evolutionary sequence comparisons to map where and the kinase domain (the surface opposite the activation loop), how the PHTH domain affects the Bruton’s tyrosine kinase (BTK) stabilizing the autoinhibited form of the kinase in a manner similar domain. -

Mechanism of Resilin Elasticity
ARTICLE Received 24 Oct 2011 | Accepted 11 Jul 2012 | Published 14 Aug 2012 DOI: 10.1038/ncomms2004 Mechanism of resilin elasticity Guokui Qin1,*, Xiao Hu1,*, Peggy Cebe2 & David L. Kaplan1 Resilin is critical in the flight and jumping systems of insects as a polymeric rubber-like protein with outstanding elasticity. However, insight into the underlying molecular mechanisms responsible for resilin elasticity remains undefined. Here we report the structure and function of resilin from Drosophila CG15920. A reversible beta-turn transition was identified in the peptide encoded by exon III and for full-length resilin during energy input and release, features that correlate to the rapid deformation of resilin during functions in vivo. Micellar structures and nanoporous patterns formed after beta-turn structures were present via changes in either the thermal or the mechanical inputs. A model is proposed to explain the super elasticity and energy conversion mechanisms of resilin, providing important insight into structure–function relationships for this protein. Furthermore, this model offers a view of elastomeric proteins in general where beta-turn-related structures serve as fundamental units of the structure and elasticity. 1 Department of Biomedical Engineering, Tufts University, Medford, Massachusetts 02155, USA. 2 Department of Physics and Astronomy, Tufts University, Medford, Massachusetts 02155, USA. *These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.L.K. (email: [email protected]). -
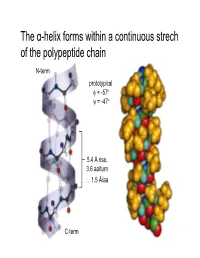
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Pleckstrin Homology Domains and the Cytoskeleton
FEBS 25627 FEBS Letters 513 (2002) 71^76 View metadata, citation and similar papers at core.ac.uk brought to you by CORE Minireview provided by Elsevier - Publisher Connector Pleckstrin homology domains and the cytoskeleton Mark A. Lemmona;Ã, Kathryn M. Fergusona, Charles S. Abramsb;Ã aDepartment of Biochemistry and Biophysics, University of Pennsylvania School of Medicine, 809C Stellar-Chance Laboratories, 422 Curie Boulevard, Philadelphia, PA 19104-6059, USA bDepartment of Medicine, University of Pennsylvania School of Medicine, 912 BRB II/III, 421 Curie Boulevard, Philadelphia, PA 19104-6160, USA Received 20 October 2001; revised 30 October 2001; accepted 14 November 2001 First published online 6 December 2001 Edited by Gianni Cesareni and Mario Gimona some 27 di¡erent proteins contain a total of 36 PH domains, Abstract Pleckstrin homology (PH) domains are 100^120 amino acid protein modules best known for their ability to bind making the PH domain the 17th most common yeast domain phosphoinositides. All possess an identical core L-sandwich fold [6]. The sequence characteristics used to identify PH domains and display marked electrostatic sidedness. The binding site for appear to de¢ne a particular protein fold that has now been phosphoinositides lies in the center of the positively charged face. seen in the X-ray crystal structures and/or nuclear magnetic In some cases this binding site is well defined, allowing highly resonance (NMR) structures of some 13 di¡erent PH domains specific and strong ligand binding. In several of these cases the [7^12]. Each of these PH domains possesses an almost iden- PH domains specifically recognize 3-phosphorylated phospho- tical core L-sandwich structure (described below), despite pair- inositides, allowing them to drive membrane recruitment in wise sequence identities between PH domains that range from response to phosphatidylinositol 3-kinase activation. -

Modeling and Predicting Super-Secondary Structures of Transmembrane Beta-Barrel Proteins Thuong Van Du Tran
Modeling and predicting super-secondary structures of transmembrane beta-barrel proteins Thuong van Du Tran To cite this version: Thuong van Du Tran. Modeling and predicting super-secondary structures of transmembrane beta-barrel proteins. Bioinformatics [q-bio.QM]. Ecole Polytechnique X, 2011. English. NNT : 2011EPXX0104. pastel-00711285 HAL Id: pastel-00711285 https://pastel.archives-ouvertes.fr/pastel-00711285 Submitted on 23 Jun 2012 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THESE` pr´esent´ee pour obtenir le grade de DOCTEUR DE L’ECOLE´ POLYTECHNIQUE Sp´ecialit´e: INFORMATIQUE par Thuong Van Du TRAN Titre de la th`ese: Modeling and Predicting Super-secondary Structures of Transmembrane β-barrel Proteins Soutenue le 7 d´ecembre 2011 devant le jury compos´ede: MM. Laurent MOUCHARD Rapporteurs Mikhail A. ROYTBERG MM. Gregory KUCHEROV Examinateurs Mireille REGNIER M. Jean-Marc STEYAERT Directeur Laboratoire d’Informatique UMR X-CNRS 7161 Ecole´ Polytechnique, 91128 Plaiseau CEDEX, FRANCE Composed with LATEX !c Thuong Van Du Tran. All rights reserved. Contents Introduction 1 1Fundamentalreviewofproteins 5 1.1 Introduction................................... 5 1.2 Proteins..................................... 5 1.2.1 Aminoacids............................... 5 1.2.2 Properties of amino acids . -

Development and Characterization of Novel Bioluminescent Reporters of Cellular Activity by Derrick C. Cumberbatch Dissertation
Development and Characterization of Novel Bioluminescent Reporters of Cellular Activity By Derrick C. Cumberbatch Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biological Sciences May 10, 2019 Nashville, Tennessee Approved: C. David Weaver, Ph.D. Douglas McMahon, Ph.D. Qi Zhang, Ph.D. Carl Johnson, Ph.D. To my beloved and supportive wife Alicia, and to my parents Cameron and Marcia Cumberbatch. ii ACKNOWLEDGEMENTS This work was made possible by financial support from the NIMH grants MH107713 and MH116150 awarded to Carl Johnson, Ph.D. as well as funds provided by the Vanderbilt University Dissertation Enhancement Grant, graciously awarded to me by the Graduate School. I appreciate Dr. Carl Johnson for taking me into his lab and providing me with ample tools that aided in the successful completion of my Ph.D. I would like to express gratitude to my committee members Drs. David Weaver, Douglas McMahon, Qi Zhang, and the late Dr. Donna Webb for guiding me through the process of becoming a competent researcher. I would also like to make special mention of Jie Yang, Ph.D. whose persistent efforts and sage advice were an ever-present help during my graduate studies. His one-on-one training provided me with many skills that will serve me well as a molecular biologist. Meaningful contributions from the other current and past members of the Johnson lab group, Yao Xu, Ph.D., Tetsuya Mori, Ph.D., Shuqun Shi, Ph.D., Chi Zhao, Ph.D., Peijun Ma, Ph.D., He Huang, Ph.D., Kathryn Campbell, Briana Wyzinski, Kevin Kelly, Maria Luisa Jabbur, Carla O’Neale and Ian Dew deserve to be highlighted here as well.