Using Molecular Constraints and Unnatural Amino Acids to Manipulate and Interrogate Protein Structure, Dynamics, and Self- Assembly
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanism of Resilin Elasticity
ARTICLE Received 24 Oct 2011 | Accepted 11 Jul 2012 | Published 14 Aug 2012 DOI: 10.1038/ncomms2004 Mechanism of resilin elasticity Guokui Qin1,*, Xiao Hu1,*, Peggy Cebe2 & David L. Kaplan1 Resilin is critical in the flight and jumping systems of insects as a polymeric rubber-like protein with outstanding elasticity. However, insight into the underlying molecular mechanisms responsible for resilin elasticity remains undefined. Here we report the structure and function of resilin from Drosophila CG15920. A reversible beta-turn transition was identified in the peptide encoded by exon III and for full-length resilin during energy input and release, features that correlate to the rapid deformation of resilin during functions in vivo. Micellar structures and nanoporous patterns formed after beta-turn structures were present via changes in either the thermal or the mechanical inputs. A model is proposed to explain the super elasticity and energy conversion mechanisms of resilin, providing important insight into structure–function relationships for this protein. Furthermore, this model offers a view of elastomeric proteins in general where beta-turn-related structures serve as fundamental units of the structure and elasticity. 1 Department of Biomedical Engineering, Tufts University, Medford, Massachusetts 02155, USA. 2 Department of Physics and Astronomy, Tufts University, Medford, Massachusetts 02155, USA. *These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.L.K. (email: [email protected]). -
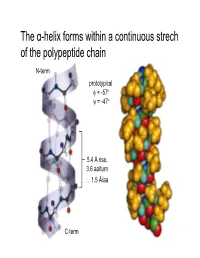
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Synthesis and Nmr Studies of a Β-Turn Mimetic Molecular
SYNTHESIS AND NMR STUDIES OF A β-TURN MIMETIC MOLECULAR TORSION BALANCE by Melissa Ann Liberatore B.S. Chemistry, Lehigh University, 2006 Submitted to the Graduate Faculty of the Kenneth P. Dietrich School of Arts and Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2012 UNIVERSITY OF PITTSBURGH DIETRICH SCHOOL OF ARTS AND SCIENCES This dissertation was presented by Melissa Ann Liberatore It was defended on July 23, 2012 and approved by Professor Dennis Curran, Department of Chemistry Professor Michael Trakselis, Department of Chemistry Professor Judith Klein-Seetharaman, Department of Structural Biology Dissertation Advisor: Professor Craig Wilcox, Department of Chemistry ii Copyright © by Melissa Ann Liberatore 2012 iii SYNTHESIS AND NMR STUDIES OF A β-TURN MIMETIC MOLECULAR TORSION BALANCE Melissa Ann Liberatore, PhD University of Pittsburgh, 2012 The attainment of precise measurements of the molecular forces that influence protein folding is important in order to further understand peptide dynamics and stability. A hybrid synthetic- natural peptide motif, combining an o,o,o’-trisubstituted biphenyl with an (ortho-tolyl)-amide, was synthesized in multiple formats and studied by NMR to probe the effects of amino acid substitutions on antiparallel beta-sheet configuration and stability. The potential of this “molecular torsion balance” as a beta-turn mimic was demonstrated by quantifying the rotational barriers about several axes. The free-energy rotational barrier of the aryl-aryl bond was found to be 35.7 kcal mol-1 at 418 K in hexanes. EXSY analysis was also used to measure barriers about -1 -1 the N-aryl (20.9 kcal mol at 343 K in toluene-d8) and N-CO bonds (17.2 kcal mol at 298 K in chloroform-d). -

Duke University Dissertation Template
Order and Disorder in Protein Biomaterial Design by Stefan Daniel Roberts Department of Biomedical Engineering Duke University Date:_______________________ Approved: ___________________________ Ashutosh Chilkoti, Supervisor ___________________________ Joel Collier ___________________________ Brenton Hoffman ___________________________ Terrence Oas ___________________________ Rohit Pappu Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biomedical Engineering in the Graduate School of Duke University 2018 ABSTRACT Order and Disorder in Protein Biomaterial Design by Stefan Daniel Roberts Department of Biomedical Engineering Duke University Date:_______________________ Approved: ___________________________ Ashutosh Chilkoti, Supervisor ___________________________ Joel Collier ___________________________ Brenton Hoffman ___________________________ Terrence Oas ___________________________ Rohit Pappu An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biomedical Engineering in the Graduate School of Duke University 2018 Copyright by Stefan Daniel Roberts 2018 Abstract Crystalline and amorphous materials have been extensively studied for their interesting properties, but they comprise a very small portion of the total materials space. The properties of most materials are a consequence of the interactions between their ordered and disordered components. This phenomenon is especially -

Fibrillar Self-Assembly of a Chimeric Elastin-Resilin Inspired Engineered Polypeptide
nanomaterials Article Fibrillar Self-Assembly of a Chimeric Elastin-Resilin Inspired Engineered Polypeptide Angelo Bracalello 1, Valeria Secchi 2,* , Roberta Mastrantonio 2, Antonietta Pepe 1, Tiziana Persichini 2, Giovanna Iucci 2 , Brigida Bochicchio 1,* and Chiara Battocchio 2,* 1 Department of Sciences, University of Basilicata, Via Ateneo Lucano, 10, 85100 Potenza, Italy; [email protected] (A.B.); [email protected] (A.P.) 2 Department of Sciences, University of Roma Tre, Via della Vasca Navale, 79, 00146 Rome, Italy; [email protected] (R.M.); [email protected] (T.P.); [email protected] (G.I.) * Correspondence: [email protected] (V.S.); [email protected] (B.B.); [email protected] (C.B.); Tel.: +39-06-5733-3400 (C.B.) Received: 2 October 2019; Accepted: 11 November 2019; Published: 14 November 2019 Abstract: In the field of tissue engineering, recombinant protein-based biomaterials made up of block polypeptides with tunable properties arising from the functionalities of the individual domains are appealing candidates for the construction of medical devices. In this work, we focused our attention on the preparation and structural characterization of nanofibers from a chimeric-polypeptide-containing resilin and elastin domain, designed on purpose to enhance its cell-binding ability by introducing a specific fibronectin-derived Arg-Gly-Asp (RGD) sequence. The polypeptide ability to self-assemble was investigated. The molecular and supramolecular structure was characterized by Scanning Electronic Microscopy (SEM) and Atomic Force Microscopy (AFM), circular dichroism, state-of-the-art synchrotron radiation-induced techniques X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure spectroscopy (NEXAFS). -

Pi-Pi Contacts Are an Overlooked Protein Feature Relevant to Phase
RESEARCH ARTICLE Pi-Pi contacts are an overlooked protein feature relevant to phase separation Robert McCoy Vernon1, Paul Andrew Chong1, Brian Tsang1,2, Tae Hun Kim1, Alaji Bah1†, Patrick Farber1‡, Hong Lin1, Julie Deborah Forman-Kay1,2* 1Program in Molecular Medicine, Hospital for Sick Children, Toronto, Canada; 2Department of Biochemistry, University of Toronto, Toronto, Canada Abstract Protein phase separation is implicated in formation of membraneless organelles, signaling puncta and the nuclear pore. Multivalent interactions of modular binding domains and their target motifs can drive phase separation. However, forces promoting the more common phase separation of intrinsically disordered regions are less understood, with suggested roles for multivalent cation-pi, pi-pi, and charge interactions and the hydrophobic effect. Known phase- separating proteins are enriched in pi-orbital containing residues and thus we analyzed pi- interactions in folded proteins. We found that pi-pi interactions involving non-aromatic groups are widespread, underestimated by force-fields used in structure calculations and correlated with solvation and lack of regular secondary structure, properties associated with disordered regions. We present a phase separation predictive algorithm based on pi interaction frequency, highlighting proteins involved in biomaterials and RNA processing. DOI: https://doi.org/10.7554/eLife.31486.001 *For correspondence: [email protected] Introduction † Present address: Department Protein phase separation has important implications for cellular organization and signaling of Biochemistry and Molecular (Mitrea and Kriwacki, 2016; Brangwynne et al., 2009; Su et al., 2016), RNA processing Biology, SUNY Upstate Medical University, New York, United (Sfakianos et al., 2016), biological materials (Yeo et al., 2011) and pathological aggregation States; ‡Zymeworks, Vancouver, (Taylor et al., 2016). -

Elastin-Like Polypeptides As Models of Intrinsically Disordered Proteins ⇑ Stefan Roberts 1, Michael Dzuricky ,1, Ashutosh Chilkoti
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 589 (2015) 2477–2486 journal homepage: www.FEBSLetters.org Review Elastin-like polypeptides as models of intrinsically disordered proteins ⇑ Stefan Roberts 1, Michael Dzuricky ,1, Ashutosh Chilkoti Duke University, Durham, NC, United States article info abstract Article history: Elastin-like polypeptides (ELPs) are a class of stimuli-responsive biopolymers inspired by the intrin- Received 13 July 2015 sically disordered domains of tropoelastin that are composed of repeats of the VPGXG pentapeptide Revised 18 August 2015 motif, where X is a ‘‘guest residue”. They undergo a reversible, thermally triggered lower critical Accepted 19 August 2015 solution temperature (LCST) phase transition, which has been utilized for a variety of applications Available online 29 August 2015 including protein purification, affinity capture, immunoassays, and drug delivery. ELPs have been Edited by Wilhelm Just extensively studied as protein polymers and as biomaterials, but their relationship to other disordered proteins has heretofore not been established. The biophysical properties of ELPs that lend them their unique material behavior are similar to the properties of many intrinsically Keywords: Elastin-like polypeptide disordered proteins (IDP). Their low sequence complexity, phase behavior, and elastic properties Intrinsically disordered protein make them an interesting ‘‘minimal” artificial IDP, and the study of ELPs can hence provide insights Phase transition into the behavior of other more complex IDPs. Motivated by this emerging realization of the Biopolymer similarities between ELPs and IDPs, this review discusses the biophysical properties of ELPs, their Tandem repeat biomedical utility, and their relationship to other disordered polypeptide sequences. -

The Structure of Small Beta Barrels
bioRxiv preprint doi: https://doi.org/10.1101/140376; this version posted May 22, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Structure of Small Beta Barrels Philippe Youkharibache*, Stella Veretnik1, Qingliang Li, Philip E. Bourne*1 National Center for Biotechnology Information, The National Library of Medicine, The National Institutes of Health, Bethesda Maryland 20894 USA. *To whom correspondence should be addressed at [email protected] and [email protected] 1 Current address: Department of Biomedical Engineering, The University of Virginia, Charlottesville VA 22908 USA. 1 bioRxiv preprint doi: https://doi.org/10.1101/140376; this version posted May 22, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract The small beta barrel is a protein structural domain, highly conserved throughout evolution and hence exhibits a broad diversity of functions. Here we undertake a comprehensive review of the structural features of this domain. We begin with what characterizes the structure and the variable nomenclature that has been used to describe it. We then go on to explore the anatomy of the structure and how functional diversity is achieved, including through establishing a variety of multimeric states, which, if misformed, contribute to disease states. -

Dynamics in Folded and Unfolded Peptides and Proteins Measured by Triplet-Triplet Energy Transfer
Fakultät für Chemie Lehrstuhl für Biophysikalische Chemie Dynamics in Folded and Unfolded Peptides and Proteins Measured by Triplet-Triplet Energy Transfer Natalie D. Merk Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. Aymelt Itzen Prüfer der Dissertation: 1. Univ.-Prof. Dr. Thomas Kiefhaber (Martin-Luther-Universität Halle-Wittenberg) 2. Univ.-Prof. Dr. Michael Groll Die Dissertation würde am 09.02.2015 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 05.03.2015 angenommen. ii Content Introduction 1 1.1 Proteins 1 1.2 Protein folding 1 1.2.1 The unfolded state of proteins 2 1.2.2 The native state of proteins 5 1.2.3 Protein stability 6 1.2.4 Barriers in protein folding 7 1.2.5 The effect of friction on protein folding kinetics 10 1.3 Triplet-triplet energy transfer as suitable tool to study protein folding 12 1.3.1 Loop formation in polypeptide chains measured by TTET 14 1.4 Turns in peptides and proteins 19 1.4.1 The role of β-turns in protein folding 20 1.5 Carp β-parvalbumin as an appropriate model to study protein folding by TTET 23 1.5.1 Site-specific modification of proteins via incorporation of unnatural amino acids and click chemistry 25 Aim of Research 29 Material and Methods 33 3.1 Used materials 33 3.2 Solid-phase peptide synthesis (SPPS) 33 3.3 Peptide modification 34 3.3.1 Introduction of chromophores for triplet-triplet -

Supplementary Materials: Understanding Insulin
Supplementary Materials: Understanding Insulin Endocrinology in Decapod Crustacea: Molecular Modelling Characterization of an Insulin-Binding Protein and Insulin-Like Peptides in the Eastern Spiny Lobster, Sagmariasus verreauxi Figure S1: Modelling criteria for (a) Sv-IGFBP; (b) Cq-IGFBP_N’; (c) Sv-IAG; (d) Sv-ILP1; (e) Sv-ILP2. In Alpha helix (Hh) each case the trimmed modelled sequence is given 310 helix (Gg) (subscript numbering in brackets indicating the native Pi helix (Ii) amino acid position) with the following associated details Beta bridge (Bb) listed: (i) model templates; (ii) input sequence alignments; Extended strand (Ee) (iii) secondary structure predictions and resulting Beta turn (Tt) additional restraints (implemented due to deviation from Bend region (Ss) the chosen template), also shown in sequence in red bold; Random coil (Cc) and (iv) defined bonds and any additional refinement. Removed in refinement (strike) Key as shown, colouration as previously described. (a) Sv-IGFBP (21)1QDVVTECGECDRSNCPEVKTCLGGKVQDACGCCEVCALGLGQRCDSQDSGDSTDYGSCGEYLVCRTRTDTG GTDEATCVCENPDPVCGSDGITYSTLCHLLQETTEKPDVFVAVRGPCKGVPVIKSKPEDKIRPLGSILVLDCEAA GYPVPEITWELNRPDGSTMKLPGDDSSFAVQVRGGPEDHMVTGWVQIMRITKKSLGIYTCVATNTEGETRVSATV ALKEHGEKEDSMNKL236(256) (i) Model template selections Insulin-binding + kazal domains: HTRA1 (3TJQ_A) Immunoglobulin domain: Myosin light chain kinase (2CQV_A) (ii) Input sequence alignments Sv-IGFBP 1 QDVVTECG-ECDRSNCPEV-KTCLGGKVQDACGCCEVCALGLGQRCDSQDSGDSTDYGSCGEYLVCRTRT 68 3TJQ_A 13 FQGSAGCPDRCEPARCPPQPEHCEGGRARDACGCCEVCGAPEGAACGLQ-------EGPCGEGLQCVVPF -

From Sequence to Structure
1 From Sequence to Structure The genomics revolution is providing gene sequences in exponentially increasing numbers. Converting this sequence information into functional information for the gene products coded by these sequences is the challenge for post-genomic biology. The first step in this process will often be the interpretation of a protein sequence in terms of the three- dimensional structure into which it folds. This chapter summarizes the basic concepts that underlie the relationship between sequence and structure and provides an overview of the architecture of proteins. 1-0 Overview: Protein Function and Architecture 1-1 Amino Acids 1-2 Genes and Proteins 1-3 The Peptide Bond 1-4 Bonds that Stabilize Folded Proteins 1-5 Importance and Determinants of Secondary Structure 1-6 Properties of the Alpha Helix 1-7 Properties of the Beta Sheet 1-8 Prediction of Secondary Structure 1-9 Folding 1-10 Tertiary Structure 1-11 Membrane Protein Structure 1-12 Protein Stability: Weak Interactions and Flexibility 1-13 Protein Stability: Post-Translational Modifications 1-14 The Protein Domain 1-15 The Universe of Protein Structures 1-16 Protein Motifs 1-17 Alpha Domains and Beta Domains 1-18 Alpha/Beta, Alpha+Beta and Cross-Linked Domains 1-19 Quaternary Structure: General Principles 1-20 Quaternary Structure: Intermolecular Interfaces 1-21 Quaternary Structure: Geometry 1-22 Protein Flexibility 1-0 Overview: Protein Function and Architecture Binding TATA binding protein Myoglobin Specific recognition of other molecules is central to protein function. The molecule that is bound (the ligand) can be as small as the oxygen molecule that coordinates to the heme group of myoglobin, or as large as the specific DNA sequence (called the TATA box) that is bound—and distorted—by the TATA binding protein. -

Beta Turn Propensity and Polymer Scaling Exponent Identify Intrinsically Disordered Proteins 2 That Phase Separate 3 4 5 6 Elisia A
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189613; this version posted July 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Beta Turn Propensity and Polymer Scaling Exponent Identify Intrinsically Disordered Proteins 2 that Phase Separate 3 4 5 6 Elisia A. Paiz,1 John J. Correia,2 Nicholas C. Fitzkee,3 Loren E. Hough,4,5 and Steven T. Whitten1* 7 8 9 1 Department of Chemistry and Biochemistry, Texas State University, San Marcos, Texas 78666, United States 10 2 Department of Cell and Molecular Biology, University of Mississippi Medical Center, Jackson, MS 39211, United 11 States 12 3 Department of Chemistry, Mississippi State University, Mississippi State, Mississippi 39762, United States 13 4 Department of Physics, University of Colorado Boulder, Boulder CO 80309, United States 14 5 BioFrontiers Institute, University of Colorado Boulder, Boulder CO 80309, United States 15 16 17 Abstract 18 19 The complex cellular milieu can spontaneously de-mix in a process driven in part by proteins that are 20 intrinsically disordered (ID). We hypothesized that protein self-interactions that determine the polymer 21 scaling exponent, v, of monomeric ID proteins (IDPs), also facilitate de-mixing transitions into phase 22 separated assemblies. We analyzed a protein database containing subsets that are folded, ID, or IDPs 23 identified previously to spontaneously phase separate. We found that the subsets differentiated into distinct 24 protein classes according to sequence-based calculations of v and, surprisingly, the propensity in the 25 sequence for adopting the β-turn.