Pi-Pi Contacts Are an Overlooked Protein Feature Relevant to Phase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

2020 Program Book
PROGRAM BOOK Note that TAGC was cancelled and held online with a different schedule and program. This document serves as a record of the original program designed for the in-person meeting. April 22–26, 2020 Gaylord National Resort & Convention Center Metro Washington, DC TABLE OF CONTENTS About the GSA ........................................................................................................................................................ 3 Conference Organizers ...........................................................................................................................................4 General Information ...............................................................................................................................................7 Mobile App ....................................................................................................................................................7 Registration, Badges, and Pre-ordered T-shirts .............................................................................................7 Oral Presenters: Speaker Ready Room - Camellia 4.......................................................................................7 Poster Sessions and Exhibits - Prince George’s Exhibition Hall ......................................................................7 GSA Central - Booth 520 ................................................................................................................................8 Internet Access ..............................................................................................................................................8 -

DEAD-Box RNA Helicases in Cell Cycle Control and Clinical Therapy
cells Review DEAD-Box RNA Helicases in Cell Cycle Control and Clinical Therapy Lu Zhang 1,2 and Xiaogang Li 2,3,* 1 Department of Nephrology, Renmin Hospital of Wuhan University, Wuhan 430060, China; [email protected] 2 Department of Internal Medicine, Mayo Clinic, 200 1st Street, SW, Rochester, MN 55905, USA 3 Department of Biochemistry and Molecular Biology, Mayo Clinic, 200 1st Street, SW, Rochester, MN 55905, USA * Correspondence: [email protected]; Tel.: +1-507-266-0110 Abstract: Cell cycle is regulated through numerous signaling pathways that determine whether cells will proliferate, remain quiescent, arrest, or undergo apoptosis. Abnormal cell cycle regula- tion has been linked to many diseases. Thus, there is an urgent need to understand the diverse molecular mechanisms of how the cell cycle is controlled. RNA helicases constitute a large family of proteins with functions in all aspects of RNA metabolism, including unwinding or annealing of RNA molecules to regulate pre-mRNA, rRNA and miRNA processing, clamping protein complexes on RNA, or remodeling ribonucleoprotein complexes, to regulate gene expression. RNA helicases also regulate the activity of specific proteins through direct interaction. Abnormal expression of RNA helicases has been associated with different diseases, including cancer, neurological disorders, aging, and autosomal dominant polycystic kidney disease (ADPKD) via regulation of a diverse range of cellular processes such as cell proliferation, cell cycle arrest, and apoptosis. Recent studies showed that RNA helicases participate in the regulation of the cell cycle progression at each cell cycle phase, including G1-S transition, S phase, G2-M transition, mitosis, and cytokinesis. -

R-Loop Proximity Proteomics Identifies a Role of DDX41 in Transcription- 2 Associated Genomic Instability
R-loop proximity proteomics identies a role of DDX41 in transcription-associated genomic instability Thorsten Mosler Institute of Molecular Biology (IMB) Francesca Conte Institute of Molecular Biology (IMB) Ivan Mikicic Institute of Molecular Biology (IMB) https://orcid.org/0000-0003-3393-0688 Nastasja Kreim Institute of Molecular Biology Martin Möckel Institute of Molecular Biology (IMB) Johanna Flach Medical Faculty Mannheim of the Heidelberg University Brian Luke Johannes Gutenberg University of Mainz https://orcid.org/0000-0002-1648-5511 Petra Beli ( [email protected] ) Institute of Molecular Biology (IMB) https://orcid.org/0000-0001-9507-9820 Article Keywords: DDX41, genomic instability, proximity proteomics, R-loops, transcription Posted Date: April 26th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-337351/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License 1 R-loop proximity proteomics identifies a role of DDX41 in transcription- 2 associated genomic instability 1 1 1 1 1 3 Thorsten Mosler , Francesca Conte , Ivan Mikicic , Nastasja Kreim , Martin M. Möckel , 4 Johanna Flach2, Brian Luke1,3, Petra Beli1,3,# 5 6 1Institute of Molecular Biology (IMB), Mainz, Germany 7 2Department of Hematology and Oncology, Medical Faculty Mannheim of the Heidelberg 8 University, Mannheim, Germany 9 3Institute of Developmental Biology and Neurobiology (IDN), Johannes Gutenberg-Universität, 10 Mainz, Germany. 11 #Correspondence should be addressed to [email protected] 12 13 14 15 16 17 18 19 20 21 22 Keywords: DDX41, genomic instability, proximity proteomics, R-loops, transcription 23 1 24 Abstract 25 Transcription can pose a threat to genomic stability through the formation of R-loops that 26 obstruct the progression of replication forks. -

Mechanism of Resilin Elasticity
ARTICLE Received 24 Oct 2011 | Accepted 11 Jul 2012 | Published 14 Aug 2012 DOI: 10.1038/ncomms2004 Mechanism of resilin elasticity Guokui Qin1,*, Xiao Hu1,*, Peggy Cebe2 & David L. Kaplan1 Resilin is critical in the flight and jumping systems of insects as a polymeric rubber-like protein with outstanding elasticity. However, insight into the underlying molecular mechanisms responsible for resilin elasticity remains undefined. Here we report the structure and function of resilin from Drosophila CG15920. A reversible beta-turn transition was identified in the peptide encoded by exon III and for full-length resilin during energy input and release, features that correlate to the rapid deformation of resilin during functions in vivo. Micellar structures and nanoporous patterns formed after beta-turn structures were present via changes in either the thermal or the mechanical inputs. A model is proposed to explain the super elasticity and energy conversion mechanisms of resilin, providing important insight into structure–function relationships for this protein. Furthermore, this model offers a view of elastomeric proteins in general where beta-turn-related structures serve as fundamental units of the structure and elasticity. 1 Department of Biomedical Engineering, Tufts University, Medford, Massachusetts 02155, USA. 2 Department of Physics and Astronomy, Tufts University, Medford, Massachusetts 02155, USA. *These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.L.K. (email: [email protected]). -
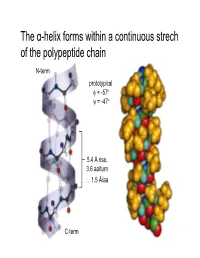
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Download Article (PDF)
Article in press - uncorrected proof BioMol Concepts, Vol. 2 (2011), pp. 343–352 • Copyright ᮊ by Walter de Gruyter • Berlin • Boston. DOI 10.1515/BMC.2011.033 Review Fragile X family members have important and non-overlapping functions Claudia Winograd2 and Stephanie Ceman1,2,* member, FMR1, was isolated by positional cloning of the X 1 Department of Cell and Developmental Biology, chromosomal region containing the inducible fragile site in University of Illinois, 601 S. Goodwin Avenue, individuals with fragile X syndrome (1). Cloning of the gene Urbana–Champaign, IL 61801, USA revealed the molecular defect to be a trinucleotide (CGG) 2 Neuroscience Program and College of Medicine, repeat expansion in exon 1 (2). Normally, individuals have University of Illinois, 601 S. Goodwin Avenue, less than 45 repeats with an average around 30 repeats; how- Urbana–Champaign, IL 61801, USA ever, expansion to greater than 200 repeats leads to aberrant methylation of the cytosines, leading to recruitment of * Corresponding author histone deacetylases with consequent transcriptional silenc- e-mail: [email protected] ing of the FMR1 locus (3). Thus, individuals with fragile X syndrome do not express transcript from the FMR1 locus. Abstract To identify the Xenopus laevis ortholog of FMR1 for further use in developmental studies, the human FMR1 gene was The fragile X family of genes encodes a small family of used to screen a cDNA library prepared from Xenopus laevis RNA binding proteins including FMRP, FXR1P and FXR2P ovary. In addition to identifying the Xenopus laevis ortholog that were identified in the 1990s. All three members are of FMR1, the first autosomal paralog FXR1 was discovered encoded by 17 exons and show alternative splicing at the 39 because of its sequence similarity to FMR1 (4). -

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . DDX41 (N-13): sc-104868 BACKGROUND RECOMMENDED SECONDARY REAGENTS DDX41 (probable ATP-dependent RNA helicase DDX41, DEAD-box protein To ensure optimal results, the following support (secondary) reagents are abstrakt homolog) is a 622 amino acid protein encoded by the human gene recommended: 1) Western Blotting: use donkey anti-goat IgG-HRP: sc-2020 DDX41. DDX41 belongs to the DEAD-box helicase family (DDX41 subfamily) (dilution range: 1:2000-1:100,000) or Cruz Marker™ compatible donkey and contains one CCHC-type zinc finger, one helicase ATP-binding domain and anti- goat IgG-HRP: sc-2033 (dilution range: 1:2000-1:5000), Cruz Marker™ one helicase C-terminal domain. DDX41 is required during post-transcriptional Molecular Weight Standards: sc-2035, TBS Blotto A Blocking Reagent: gene expression and is thought to be involved in pre-mRNA splicing. DDX41 sc-2333 and Western Blotting Luminol Reagent: sc-2048. 2) Immunoprecip- is believed to be a probable ATP-dependent RNA helicase. RNA helicases are itation: use Protein A/G PLUS-Agarose: sc-2003 (0.5 ml agarose/2.0 ml). highly conserved enzymes that utilize the energy derived from NTP hydrolysis 3) Immunofluorescence: use donkey anti-goat IgG-FITC: sc-2024 (dilution to modulate the structure of RNA. RNA helicases participate in all biological range: 1:100-1:400) or donkey anti-goat IgG-TR: sc-2783 (dilution range: processes that involve RNA, including transcription, splicing and translation. 1:100-1:400) with UltraCruz™ Mounting Medium: sc-24941. REFERENCES DATA 1. Irion, U. -

Dysregulated RNA-Induced Silencing Complex (RISC) Assembly Within CNS Corresponds with Abnormal Mirna Expression During Autoimmune Demyelination
The Journal of Neuroscience, May 13, 2015 • 35(19):7521–7537 • 7521 Neurobiology of Disease Dysregulated RNA-Induced Silencing Complex (RISC) Assembly within CNS Corresponds with Abnormal miRNA Expression during Autoimmune Demyelination Przemysław Lewkowicz, Hanna Cwiklin´ska, Marcin P. Mycko, Maria Cichalewska, Małgorzata Domowicz, Natalia Lewkowicz, Anna Jurewicz, and Krzysztof W. Selmaj Department of Neurology, Laboratory of Neuroimmunology, Medical University of Lodz, 92-213 Lodz, Poland MicroRNAs (miRNAs) associate with Argonaute (Ago), GW182, and FXR1 proteins to form RNA-induced silencing complexes (RISCs). RISCs represent a critical checkpoint in the regulation and bioavailability of miRNAs. Recent studies have revealed dysregulation of miRNAs in multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE); however, the function of RISCs in EAE and MS is largely unknown. Here, we examined the expression of Ago, GW182, and FXR1 in CNS tissue, oligodendrocytes (OLs), brain-infiltrating T lymphocytes, and CD3 ϩsplenocytes (SCs) of EAE mic, and found that global RISC protein levels were signifi- cantly dysregulated. Specifically, Ago2 and FXR1 levels were decreased in OLs and brain-infiltrating T cells in EAE mice. Accordingly, assembly of Ago2/GW182/FXR1 complexes in EAE brain tissues was disrupted, as confirmed by immunoprecipitation experiments. In parallel with alterations in RISC complex content in OLs, we found downregulation of miRNAs essential for differentiation and survival of OLs and myelin synthesis. In brain-infiltrating T lymphocytes, aberrant RISC formation contributed to miRNA-dependent proinflam- matory helper T-cell polarization. In CD3 ϩ SCs, we found increased expression of both Ago2 and FXR1 in EAE compared with nonim- munized mice. -

TAF15 Is Important for Cellular Proliferation and Regulates the Expression of a Subset of Cell Cycle Genes Through Mirnas
Oncogene (2013) 32, 4646–4655 & 2013 Macmillan Publishers Limited All rights reserved 0950-9232/13 www.nature.com/onc ORIGINAL ARTICLE TAF15 is important for cellular proliferation and regulates the expression of a subset of cell cycle genes through miRNAs M Ballarino1, L Jobert1,4, D Dembe´le´ 1, P de la Grange2, D Auboeuf3 and L Tora1 TAF15 (formerly TAFII68) is a member of the FET (FUS, EWS, TAF15) family of RNA- and DNA-binding proteins whose genes are frequently translocated in sarcomas. By performing global gene expression profiling, we found that TAF15 knockdown affects the expression of a large subset of genes, of which a significant percentage is involved in cell cycle and cell death. In agreement, TAF15 depletion had a growth-inhibitory effect and resulted in increased apoptosis. Among the TAF15-regulated genes, targets of microRNAs (miRNAs) generated from the onco-miR-17 locus were overrepresented, with CDKN1A/p21 being the top miRNAs- targeted gene. Interestingly, the levels of onco-miR-17 locus coded miRNAs (miR-17-5p and miR-20a) were decreased upon TAF15 depletion and shown to affect the post-transcriptional regulation of TAF15-dependent genes, such as CDKN1A/p21. Thus, our results demonstrate that TAF15 is required to regulate gene expression of cell cycle regulatory genes post-transcriptionally through a pathway involving miRNAs. The findings that high TAF15 levels are needed for rapid cellular proliferation and that endogenous TAF15 levels decrease during differentiation strongly suggest that TAF15 is a key regulator of maintaining a highly proliferative rate of cellular homeostasis. Oncogene (2013) 32, 4646–4655; doi:10.1038/onc.2012.490; published online 5 November 2012 Keywords: FUS/EWS/TAF15 (FET) proteins; miRNAs; transcription; proliferation; neuronal differentiation; neuroblastoma INTRODUCTION possible role of TAF15 in additional steps of RNA metabolism. -

Evidence of an Interaction Between FXR1 and Gsk3β Polymorphisms
European Psychiatry Evidence of an interaction between FXR1 and GSK3β www.cambridge.org/epa polymorphisms on levels of Negative Symptoms of Schizophrenia and their response to antipsychotics Research Article 1,2 1 1 1,3 Cite this article: Rampino A, Torretta S, Antonio Rampino * , Silvia Torretta , Barbara Gelao , Federica Veneziani , Gelao B, Veneziani F, Iacoviello M, Matteo Iacoviello1 , Aleksandra Marakhovskaya3, Rita Masellis1, Ileana Andriola2, Marakhovskaya A, Masellis R, Andriola I, Sportelli L, Pergola G, Minelli A, Magri C, Leonardo Sportelli1 , Giulio Pergola1,4 , Alessandra Minelli5,6 , Chiara Magri5 , Gennarelli M, Vita A, Beaulieu JM, Bertolino A, 5,6 5,7 3 Blasi G (2021). Evidence of an interaction Massimo Gennarelli , Antonio Vita , Jean Martin Beaulieu , FXR1 GSK3β between and polymorphisms on 1,2 1,2 levels of Negative Symptoms of Schizophrenia Alessandro Bertolino and Giuseppe Blasi * and their response to antipsychotics. 1 European Psychiatry, 64(1), e39, 1–9 Group of Psychiatric Neuroscience, Department of Basic Medical Sciences, Neuroscience and Sense Organs, University of https://doi.org/10.1192/j.eurpsy.2021.26 Bari Aldo Moro, Bari, Italy; 2Azienda Ospedaliero-Universitaria Consorziale Policlinico, Bari, Italy; 3Department of Pharmacology, University of Toronto, Toronto, Ontario, Canada; 4Lieber Institute for Brain Development, Johns Hopkins Received: 17 November 2020 Medical Campus, Baltimore, Maryland, USA; 5Department of Molecular and Translational Medicine, University of Revised: 08 March 2021 Brescia, Brescia, Italy; 6Genetics Unit, IRCCS Istituto Centro San Giovanni di Dio Fatebenefratelli, Brescia, Italy and Accepted: 02 April 2021 7Department of Mental Health and Addiction Services, ASST Spedali Civili of Brescia, Brescia, Italy Keywords: FXR1; GSK3β; Negative Symptoms; Abstract Schizophrenia; treatment with antipsychotics Background. -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation.