Local Protein Structures Bernard Offmann, Manoj Tyagi, Alexandre De Brevern
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Optimal Matching Distances Between Categorical Sequences: Distortion and Inferences by Permutation Juan P
St. Cloud State University theRepository at St. Cloud State Culminating Projects in Applied Statistics Department of Mathematics and Statistics 12-2013 Optimal Matching Distances between Categorical Sequences: Distortion and Inferences by Permutation Juan P. Zuluaga Follow this and additional works at: https://repository.stcloudstate.edu/stat_etds Part of the Applied Statistics Commons Recommended Citation Zuluaga, Juan P., "Optimal Matching Distances between Categorical Sequences: Distortion and Inferences by Permutation" (2013). Culminating Projects in Applied Statistics. 8. https://repository.stcloudstate.edu/stat_etds/8 This Thesis is brought to you for free and open access by the Department of Mathematics and Statistics at theRepository at St. Cloud State. It has been accepted for inclusion in Culminating Projects in Applied Statistics by an authorized administrator of theRepository at St. Cloud State. For more information, please contact [email protected]. OPTIMAL MATCHING DISTANCES BETWEEN CATEGORICAL SEQUENCES: DISTORTION AND INFERENCES BY PERMUTATION by Juan P. Zuluaga B.A. Universidad de los Andes, Colombia, 1995 A Thesis Submitted to the Graduate Faculty of St. Cloud State University in Partial Fulfillment of the Requirements for the Degree Master of Science St. Cloud, Minnesota December, 2013 This thesis submitted by Juan P. Zuluaga in partial fulfillment of the requirements for the Degree of Master of Science at St. Cloud State University is hereby approved by the final evaluation committee. Chairperson Dean School of Graduate Studies OPTIMAL MATCHING DISTANCES BETWEEN CATEGORICAL SEQUENCES: DISTORTION AND INFERENCES BY PERMUTATION Juan P. Zuluaga Sequence data (an ordered set of categorical states) is a very common type of data in Social Sciences, Genetics and Computational Linguistics. -

Protein Structure: Data Bases and Classification
Protein Structure: Data Bases and Classification Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Reference Bourne and Weissig Structural Bioinformatics Wiley, 2003 More References 1 Structural Proteins Membrane Proteins Globular Proteins 2 Terminology • Primary Structure • Secondary Structure • Tertiary Structure • Quatenary Structure • Supersecondary Structure • Domain • Fold Hierarchy of Protein Structure Helices α 3.10 π Amino acids/turn: 3.6 3.0 4.4 Frequency ~97% ~3% rare H-bonding i, i+4 i, i+3 i, i+5 3 α-helices α-helices α-helices have handedness: α-helices have a dipole: β-sheets 4 β-sheets Have a right-handed twist! β-sheets Can form higher level structures! Super Secondary Structure Motifs 5 What is a Domain? Richardson (1981): W ithin a single subunit [polypeptide chain], contiguous portions of the polypeptide chain frequently fold into compact, local semi-independent units called domains. More About Domains • Independent folding units. • Lots of within contacts, few outside. • Domains create their own hydrophobic core. • Regions usually conserved during recombination. • Different domains of the same protein can have different functions. • Domains of the same protein may or may not interact. Why Look for Domains? Domains are the currency of protein function! 6 Domain Size • Domains can be between 25 and 500 residues long. • Most are less than 200 residues. • Domains can be smaller than 50 residues, but these need to be stabilized. Examples are the zinc finger and a scorpion toxin. Two Very Small Domains A Humdinger of a Domain 7 What’s the Domain? (Part 1) What’s the Domain? (Part 2) Homology and Analogy • Homology: Similarity in characteristics resulting from shared ancestry. -

Simulations of Я-Hairpin Folding Confined to Spherical Pores Using
Simulations of -hairpin folding confined to spherical pores using distributed computing D. K. Klimov*†, D. Newfield‡, and D. Thirumalai*† *Institute for Physical Science and Technology, University of Maryland, College Park, MD 20742; and ‡Parabon Computation, 3930 Walnut Street, Suite 100, Fairfax, VA 22030-4738 Communicated by George H. Lorimer, University of Maryland, College Park, MD, April 12, 2002 (received for review December 18, 2001) 3 ϱ We report the thermodynamics and kinetics of an off-lattice Go radius of gyration of a chain Rg at D (the size of a chain in  Ӎ model -hairpin from Ig-binding protein confined to an inert bulk solution) to N according to Rg aN . If the chain is ideal, ϭ ⌬ ϭ ͞ 2 spherical pore. Confinement enhances the stability of the hairpin then 0.5 and FU RTN(a D) . Because of the reduction due to the decrease in the entropy of the unfolded state. Compared in the translational entropy, confinement also increases the free ⌬ Ͼ ⌬ ͞⌬ ϽϽ with their values in the bulk, the rates of hairpin formation increase energy of the native state, i.e., FN 0. If FN FU 1, then in the spherical pore. Surprisingly, the dependence of the rates on localization of a protein in a confined space stabilizes the native the pore radius, Rs, is nonmonotonic. The rates reach a maximum state compared with the bulk. It also follows that there is a range ͞ b Ӎ b at Rs Rg,N 1.5, where Rg,N is the radius of gyration of the folded of D values over which stability is maximized. -

Sequence Motifs, Correlations and Structural Mapping of Evolutionary
Talk overview • Sequence profiles – position specific scoring matrix • Psi-blast. Automated way to create and use sequence Sequence motifs, correlations profiles in similarity searches and structural mapping of • Sequence patterns and sequence logos evolutionary data • Bioinformatic tools which employ sequence profiles: PFAM BLOCKS PROSITE PRINTS InterPro • Correlated Mutations and structural insight • Mapping sequence data on structures: March 2011 Eran Eyal Conservations Correlations PSSM – position specific scoring matrix • A position-specific scoring matrix (PSSM) is a commonly used representation of motifs (patterns) in biological sequences • PSSM enables us to represent multiple sequence alignments as mathematical entities which we can work with. • PSSMs enables the scoring of multiple alignments with sequences, or other PSSMs. PSSM – position specific scoring matrix Assuming a string S of length n S = s1s2s3...sn If we want to score this string against our PSSM of length n (with n lines): n alignment _ score = m ∑ s j , j j=1 where m is the PSSM matrix and sj are the string elements. PSSM can also be incorporated to both dynamic programming algorithms and heuristic algorithms (like Psi-Blast). Sequence space PSI-BLAST • For a query sequence use Blast to find matching sequences. • Construct a multiple sequence alignment from the hits to find the common regions (consensus). • Use the “consensus” to search again the database, and get a new set of matching sequences • Repeat the process ! Sequence space Position-Specific-Iterated-BLAST • Intuition – substitution matrices should be specific to sites and not global. – Example: penalize alanine→glycine more in a helix •Idea – Use BLAST with high stringency to get a set of closely related sequences. -

Computational Biology Lecture 8: Substitution Matrices Saad Mneimneh
Computational Biology Lecture 8: Substitution matrices Saad Mneimneh As we have introduced last time, simple scoring schemes like +1 for a match, -1 for a mismatch and -2 for a gap are not justifiable biologically, especially for amino acid sequences (proteins). Instead, more elaborated scoring functions are used. These scores are usually obtained as a result of analyzing chemical properties and statistical data for amino acids and DNA sequences. For example, it is known that same size amino acids are more likely to be substituted by one another. Similarly, amino acids with same affinity to water are likely to serve the same purpose in some cases. On the other hand, some mutations are not acceptable (may lead to demise of the organism). PAM and BLOSUM matrices are amongst results of such analysis. We will see the techniques through which PAM and BLOSUM matrices are obtained. Substritution matrices Chemical properties of amino acids govern how the amino acids substitue one another. In principle, a substritution matrix s, where sij is used to score aligning character i with character j, should reflect the probability of two characters substituing one another. The question is how to build such a probability matrix that closely maps reality? Different strategies result in different matrices but the central idea is the same. If we go back to the concept of a high scoring segment pair, theory tells us that the alignment (ungapped) given by such a segment is governed by a limiting distribution such that ¸sij qij = pipje where: ² s is the subsitution matrix used ² qij is the probability of observing character i aligned with character j ² pi is the probability of occurrence of character i Therefore, 1 qij sij = ln ¸ pipj This formula for sij suggests a way to constrcut the matrix s. -

Mechanism of Resilin Elasticity
ARTICLE Received 24 Oct 2011 | Accepted 11 Jul 2012 | Published 14 Aug 2012 DOI: 10.1038/ncomms2004 Mechanism of resilin elasticity Guokui Qin1,*, Xiao Hu1,*, Peggy Cebe2 & David L. Kaplan1 Resilin is critical in the flight and jumping systems of insects as a polymeric rubber-like protein with outstanding elasticity. However, insight into the underlying molecular mechanisms responsible for resilin elasticity remains undefined. Here we report the structure and function of resilin from Drosophila CG15920. A reversible beta-turn transition was identified in the peptide encoded by exon III and for full-length resilin during energy input and release, features that correlate to the rapid deformation of resilin during functions in vivo. Micellar structures and nanoporous patterns formed after beta-turn structures were present via changes in either the thermal or the mechanical inputs. A model is proposed to explain the super elasticity and energy conversion mechanisms of resilin, providing important insight into structure–function relationships for this protein. Furthermore, this model offers a view of elastomeric proteins in general where beta-turn-related structures serve as fundamental units of the structure and elasticity. 1 Department of Biomedical Engineering, Tufts University, Medford, Massachusetts 02155, USA. 2 Department of Physics and Astronomy, Tufts University, Medford, Massachusetts 02155, USA. *These authors contributed equally to this work. Correspondence and requests for materials should be addressed to D.L.K. (email: [email protected]). -

Water in Protein Structure Prediction
Water in protein structure prediction Garegin A. Papoian†‡, Johan Ulander†‡§, Michael P. Eastwood†¶, Zaida Luthey-Schultenʈ, and Peter G. Wolynes†,†† †Department of Chemistry and Biochemistry and Center for Theoretical Biological Physics, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0371; and ʈDepartment of Chemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801 Contributed by Peter G. Wolynes, December 4, 2003 Proteins have evolved to use water to help guide folding. A interactions play an important role not only in binding interfaces physically motivated, nonpairwise-additive model of water-medi- but in folding of monomeric proteins. ated interactions added to a protein structure prediction Hamilto- We use the associative memory (AM) Hamiltonian molecular nian yields marked improvement in the quality of structure pre- dynamics model as a starting point (14–16). This Hamiltonian diction for larger proteins. Free energy profile analysis suggests has two principal components: general polymer physics-based that long-range water-mediated potentials guide folding and terms that are sequence independent, collectively called ‘‘back- smooth the underlying folding funnel. Analyzing simulation tra- bone,’’ and sequence-dependent knowledge-based distance- jectories gives direct evidence that water-mediated interactions dependent additive potentials, collectively denoted as AM͞C facilitate native-like packing of supersecondary structural ele- (AM͞contact). The AM part describes interactions between all ments. Long-range pairing of hydrophilic groups is an integral part pairs of residues that are separated in sequence between 3 and of protein architecture. Specific water-mediated interactions are a 12 residues. It uses a set of nonhomologous memory proteins to universal feature of biomolecular recognition landscapes in both build a funneled energy landscape by matching fragments. -

Jacquelyn S. Fetrow
Jacquelyn S. Fetrow President and Professor of Chemistry Albright College Curriculum Vitae Office of the President Work Email: [email protected] Library and Administration Building Office Phone: 610-921-7600 N. 13th and Bern Streets, P.O. Box 15234 Reading, PA 19612 Education Ph.D. Biological Chemistry, December, 1986 B.S. Biochemistry, May, 1982 Department of Biological Chemistry Albright College, Reading, PA The Pennsylvania State University College of Medicine, Hershey, PA Graduated summa cum laude Loops: A Novel Class of Protein Secondary Structure Thesis Advisor: George D. Rose Professional Experience Albright College, Reading PA President and Professor of Chemistry June 2017-present University of Richmond, Richmond, VA Provost and Vice President of Academic Affairs July 2014-December 2016 Professor of Chemistry July 2014-May 2017 Responsibilities as Provost: Chief academic administrator for all academic matters for the University of Richmond, a university with five schools (Arts and Sciences, Business, Law, Leadership, and Professional and Continuing Studies), ~400 faculty and ~3300 full-time undergraduate and graduate students; manage the ~$91.8M annual operating budget of the Academic Affairs Division, as well as endowment and gift accounts; oversee Richmond’s Bonner Center of Civic Engagement (engage.richmond.edu), Center for International Education (international.richmond.edu), Registrar (registrar.richmond.edu), Office of Institutional Effectiveness (ifx.richmond.edu), as well as other programs and staff; partner with VP -
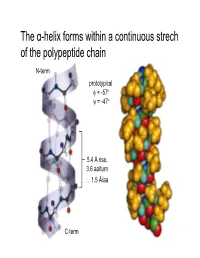
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -
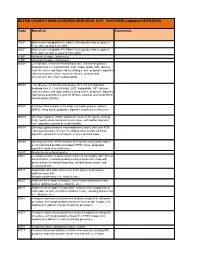
SETON CHARITY NON-COVERED SERVICES EFF: 01/01/2020 (Updated 05/05/2020)
SETON CHARITY NON-COVERED SERVICES EFF: 01/01/2020 (updated 05/05/2020) Code Narrative Comments 3051F Most recent hemoglobin A1c (HbA1c) level greater than or equal to 7.0% and less than 8.0% (DM) 3052F Most recent hemoglobin A1c (HbA1c) level greater than or equal to 8.0% and less than or equal to 9.0% (DM) 4185F Histamine therapy - intravenous 4186F Histamine therapy - intravenous 0002M Liver disease, ten biochemical assays (ALT, A2-macro- globulin, apolipoprotein A-1, total bilirubin, GGT, hapto- globin, AST, glucose, total cholesterol and triglycerides) utilizing serum, prognostic algorithm reported as quanti- tative scores for fibrosis, steatosis and alcoholic/non alco- holic steatohepatitis 0003M Liver disease, ten biochemical assays (ALT, A2-macroglobulin, apolipoprotein A-1, total bilirubin, GGT, haptoglobin, AST, glucose, total cholesterol and triglycerides) utilizing serum, prognostic algorithm reported as quantitative scores for fibrosis, steatosis and nonalcoholic steatohepatitis (NASH) 0004M Scoliosis, DNA analysis of 53 single nucleotide polymor- phisms (SNPs), using saliva, prognostic algorithm reported as a risk score 0006M Oncology (hepatic), mRNA expression levels of 161 genes, utilizing fresh hepatocellular carcinoma tumor tissue, with alpha-fetoprotein level, algorithm reported as a risk classifier 0007M Oncology (gastrointestinal neuroendocrine tumors), real- time PCR expression analysis of 51 genes, utilizing whole peripheral blood, algorithm reported as a nomogram of tumor disease index 0008M Oncology (breast), mRNA -

The Structure of Small Beta Barrels
bioRxiv preprint doi: https://doi.org/10.1101/140376; this version posted May 24, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. The Structure of Small Beta Barrels Philippe Youkharibache*, Stella Veretnik1, Qingliang Li, Philip E. Bourne*1 National Center for Biotechnology Information, The National Library of Medicine, The National Institutes of Health, Bethesda Maryland 20894 USA. *To whom correspondence should be addressed at [email protected] and [email protected] 1 Current address: Department of Biomedical Engineering, The University of Virginia, Charlottesville VA 22908 USA. 1 bioRxiv preprint doi: https://doi.org/10.1101/140376; this version posted May 24, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract The small beta barrel is a protein structural domain, highly conserved throughout evolution and hence exhibits a broad diversity of functions. Here we undertake a comprehensive review of the structural features of this domain. We begin with what characterizes the structure and the variable nomenclature that has been used to describe it. We then go on to explore the anatomy of the structure and how functional diversity is achieved, including through establishing a variety of multimeric states, which, if misformed, contribute to disease states. -

A Deep Learning Approach for Protein-Ligand Interaction Prediction
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.884841; this version posted September 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. SSnet: A Deep Learning Approach for Protein-Ligand Interaction Prediction Niraj Verma,y,x Xingming Qu,z,x Francesco Trozzi,y,x Mohamed Elsaied,{,x Nischal Karki,y,x Yunwen Tao,y,x Brian Zoltowski,y,x Eric C. Larson,z,x and Elfi Kraka∗,y,x yDepartment of Chemistry, Southern Methodist University, Dallas TX USA zDepartment of Computer Science, Southern Methodist University, Dallas TX USA {Department of Engineering Management and Information System, Southern Methodist University, Dallas TX USA xSouthern Methodist University E-mail: [email protected] Abstract Computational prediction of Protein-Ligand Interaction (PLI) is an important step in the modern drug discovery pipeline as it mitigates the cost, time, and resources re- quired to screen novel therapeutics. Deep Neural Networks (DNN) have recently shown excellent performance in PLI prediction. However, the performance is highly dependent on protein and ligand features utilized for the DNN model. Moreover, in current mod- els, the deciphering of how protein features determine the underlying principles that govern PLI is not trivial. In this work, we developed a DNN framework named SSnet that utilizes secondary structure information of proteins extracted as the curvature and torsion of the protein backbone to predict PLI. We demonstrate the performance of SSnet by comparing against a variety of currently popular machine and non-machine learning models using various metrics.