Comparative Features of Multicellular Eukaryotic Genomes (2017)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Cyanide Hydratase from Neurospora Crassa Forms a Helix Which Has a Dimeric Repeat
Appl Microbiol Biotechnol (2009) 82:271–278 DOI 10.1007/s00253-008-1735-4 BIOTECHNOLOGICALLY RELEVANT ENZYMES AND PROTEINS The cyanide hydratase from Neurospora crassa forms a helix which has a dimeric repeat Kyle C. Dent & Brandon W. Weber & Michael J. Benedik & B. Trevor Sewell Received: 2 July 2008 /Revised: 24 September 2008 /Accepted: 25 September 2008 / Published online: 23 October 2008 # Springer-Verlag 2008 Abstract The fungal cyanide hydratases form a functionally Introduction specialized subset of the nitrilases which catalyze the hydrolysis of cyanide to formamide with high specificity. The generation of cyanide containing waste results from These hold great promise for the bioremediation of cyanide cyanide utilization in a variety of applications, spanning wastes. The low resolution (3.0 nm) three-dimensional such diverse industries as gold and silver mining, metal reconstruction of negatively stained recombinant cyanide electroplating, polymer synthesis, and the production of hydratase fibers from the saprophytic fungus Neurospora fine chemicals, pharmaceuticals, dyes, and agricultural crassa by iterative helical real space reconstruction reveals products (Baxter and Cummings 2006). Traditionally, that enzyme fibers display left-handed D1 S5.4 symmetry chemical oxidation or stabilizations are used to treat these with a helical rise of 1.36 nm. This arrangement differs from wastes; however, many groups have been studying the previously characterized microbial nitrilases which demon- potential for bioremediation of such cyanide waste strate a structure built along similar principles but with a (O’Reilly and Turner 2003; Jandhyala et al. 2005). reduced helical twist. The cyanide hydratase assembly is The most promising approach utilizes cyanide degrada- stabilized by two dyadic interactions between dimers across tion by enzymes belonging to the nitrilase family (Pace and the one-start helical groove. -

Cyanide-Degrading Enzymes for Bioremediation A
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Texas A&M University CYANIDE-DEGRADING ENZYMES FOR BIOREMEDIATION A Thesis by LACY JAMEL BASILE Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE August 2008 Major Subject: Microbiology CYANIDE-DEGRADING ENZYMES FOR BIOREMEDIATION A Thesis by LACY JAMEL BASILE Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Approved by: Chair of Committee, Michael Benedik Committee Members, Susan Golden James Hu Wayne Versaw Head of Department, Vincent Cassone August 2008 Major Subject: Microbiology iii ABSTRACT Cyanide-Degrading Enzymes for Bioremediation. (August 2008) Lacy Jamel Basile, B.S., Texas A&M University Chair of Advisory Committee: Dr. Michael Benedik Cyanide-containing waste is an increasingly prevalent problem in today’s society. There are many applications that utilize cyanide, such as gold mining and electroplating, and these processes produce cyanide waste with varying conditions. Remediation of this waste is necessary to prevent contamination of soils and water. While there are a variety of processes being used, bioremediation is potentially a more cost effective alternative. A variety of fungal species are known to degrade cyanide through the action of cyanide hydratases, a specialized subset of nitrilases which hydrolyze cyanide to formamide. Here I report on previously unknown and uncharacterized nitrilases from Neurospora crassa, Gibberella zeae, and Aspergillus nidulans. Recombinant forms of four cyanide hydratases from N. -
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Structures, Functions, and Mechanisms of Filament Forming Enzymes: a Renaissance of Enzyme Filamentation
Structures, Functions, and Mechanisms of Filament Forming Enzymes: A Renaissance of Enzyme Filamentation A Review By Chad K. Park & Nancy C. Horton Department of Molecular and Cellular Biology University of Arizona Tucson, AZ 85721 N. C. Horton ([email protected], ORCID: 0000-0003-2710-8284) C. K. Park ([email protected], ORCID: 0000-0003-1089-9091) Keywords: Enzyme, Regulation, DNA binding, Nuclease, Run-On Oligomerization, self-association 1 Abstract Filament formation by non-cytoskeletal enzymes has been known for decades, yet only relatively recently has its wide-spread role in enzyme regulation and biology come to be appreciated. This comprehensive review summarizes what is known for each enzyme confirmed to form filamentous structures in vitro, and for the many that are known only to form large self-assemblies within cells. For some enzymes, studies describing both the in vitro filamentous structures and cellular self-assembly formation are also known and described. Special attention is paid to the detailed structures of each type of enzyme filament, as well as the roles the structures play in enzyme regulation and in biology. Where it is known or hypothesized, the advantages conferred by enzyme filamentation are reviewed. Finally, the similarities, differences, and comparison to the SgrAI system are also highlighted. 2 Contents INTRODUCTION…………………………………………………………..4 STRUCTURALLY CHARACTERIZED ENZYME FILAMENTS…….5 Acetyl CoA Carboxylase (ACC)……………………………………………………………………5 Phosphofructokinase (PFK)……………………………………………………………………….6 -

Changes in the Sclerotinia Sclerotiorum Transcriptome During Infection of Brassica Napus
Seifbarghi et al. BMC Genomics (2017) 18:266 DOI 10.1186/s12864-017-3642-5 RESEARCHARTICLE Open Access Changes in the Sclerotinia sclerotiorum transcriptome during infection of Brassica napus Shirin Seifbarghi1,2, M. Hossein Borhan1, Yangdou Wei2, Cathy Coutu1, Stephen J. Robinson1 and Dwayne D. Hegedus1,3* Abstract Background: Sclerotinia sclerotiorum causes stem rot in Brassica napus, which leads to lodging and severe yield losses. Although recent studies have explored significant progress in the characterization of individual S. sclerotiorum pathogenicity factors, a gap exists in profiling gene expression throughout the course of S. sclerotiorum infection on a host plant. In this study, RNA-Seq analysis was performed with focus on the events occurring through the early (1 h) to the middle (48 h) stages of infection. Results: Transcript analysis revealed the temporal pattern and amplitude of the deployment of genes associated with aspects of pathogenicity or virulence during the course of S. sclerotiorum infection on Brassica napus. These genes were categorized into eight functional groups: hydrolytic enzymes, secondary metabolites, detoxification, signaling, development, secreted effectors, oxalic acid and reactive oxygen species production. The induction patterns of nearly all of these genes agreed with their predicted functions. Principal component analysis delineated gene expression patterns that signified transitions between pathogenic phases, namely host penetration, ramification and necrotic stages, and provided evidence for the occurrence of a brief biotrophic phase soon after host penetration. Conclusions: The current observations support the notion that S. sclerotiorum deploys an array of factors and complex strategies to facilitate host colonization and mitigate host defenses. This investigation provides a broad overview of the sequential expression of virulence/pathogenicity-associated genes during infection of B. -

Eukaryotic Genome Annotation
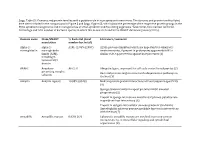
Comparative Features of Multicellular Eukaryotic Genomes (2017) (First three statistics from www.ensembl.org; other from original papers) C. elegans A. thaliana D. melanogaster M. musculus H. sapiens Species name Nematode Thale Cress Fruit Fly Mouse Human Size (Mb) 103 136 143 3,482 3,555 # Protein-coding genes 20,362 27,655 13,918 22,598 20,338 (25,498 (13,601 original (30,000 (30,000 original est.) original est.) original est.) est.) Transcripts 58,941 55,157 34,749 131,195 200,310 Gene density (#/kb) 1/5 1/4.5 1/8.8 1/83 1/97 LINE/SINE (%) 0.4 0.5 0.7 27.4 33.6 LTR (%) 0.0 4.8 1.5 9.9 8.6 DNA Elements 5.3 5.1 0.7 0.9 3.1 Total repeats 6.5 10.5 3.1 38.6 46.4 Exons % genome size 27 28.8 24.0 per gene 4.0 5.4 4.1 8.4 8.7 average size (bp) 250 506 Introns % genome size 15.6 average size (bp) 168 Arabidopsis Chromosome Structures Sorghum Whole Genome Details Characterizing the Proteome The Protein World • Sequencing has defined o Many, many proteins • How can we use this data to: o Define genes in new genomes o Look for evolutionarily related genes o Follow evolution of genes ▪ Mixing of domains to create new proteins o Uncover important subsets of genes that ▪ That deep phylogenies • Plants vs. animals • Placental vs. non-placental animals • Monocots vs. dicots plants • Common nomenclature needed o Ensure consistency of interpretations InterPro (http://www.ebi.ac.uk/interpro/) Classification of Protein Families • Intergrated documentation resource for protein super families, families, domains and functional sites o Mitchell AL, Attwood TK, Babbitt PC, et al. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Armc5 Deletion Causes Developmental Defects and Compromises T-Cell Immune Responses
ARTICLE Received 26 Jan 2016 | Accepted 4 Nov 2016 | Published 7 Feb 2017 DOI: 10.1038/ncomms13834 OPEN Armc5 deletion causes developmental defects and compromises T-cell immune responses Yan Hu 1,*, Linjiang Lao1,*, Jianning Mao1, Wei Jin1, Hongyu Luo1, Tania Charpentier2, Shijie Qi1, Junzheng Peng1, Bing Hu3, Mieczyslaw Martin Marcinkiewicz4, Alain Lamarre2 & Jiangping Wu1,5 Armadillo repeat containing 5 (ARMC5) is a cytosolic protein with no enzymatic activities. Little is known about its function and mechanisms of action, except that gene mutations are associated with risks of primary macronodular adrenal gland hyperplasia. Here we map Armc5 expression by in situ hybridization, and generate Armc5 knockout mice, which are small in body size. Armc5 knockout mice have compromised T-cell proliferation and differentiation into Th1 and Th17 cells, increased T-cell apoptosis, reduced severity of experimental autoimmune encephalitis, and defective immune responses to lymphocytic choriomeningitis virus infection. These mice also develop adrenal gland hyperplasia in old age. Yeast 2-hybrid assays identify 16 ARMC5-binding partners. Together these data indicate that ARMC5 is crucial in fetal development, T-cell function and adrenal gland growth homeostasis, and that the functions of ARMC5 probably depend on interaction with multiple signalling pathways. 1 Centre de recherche (CR), Centre hospitalier de l’Universite´ de Montre´al (CHUM), 900 Rue Saint Denis, Montre´al, Que´bec, Canada H2X 0A9. 2 Institut national de la recherche scientifique—Institut Armand-Frappier (INRS-IAF), 531 Boul. des Prairies, Laval, Que´bec, Canada H7V 1B7. 3 Anatomic Pathology, AmeriPath Central Florida, 4225 Fowler Ave. Tampa, Orlando, Florida 33617, USA. -

An Unusual Hydrophobic Core Confers Extreme Flexibility to HEAT Repeat Proteins
1596 Biophysical Journal Volume 99 September 2010 1596–1603 An Unusual Hydrophobic Core Confers Extreme Flexibility to HEAT Repeat Proteins Christian Kappel, Ulrich Zachariae, Nicole Do¨lker, and Helmut Grubmu¨ller* Department of Theoretical and Computational Biophysics, Max Planck Institute for Biophysical Chemistry, Go¨ttingen, Germany ABSTRACT Alpha-solenoid proteins are suggested to constitute highly flexible macromolecules, whose structural variability and large surface area is instrumental in many important protein-protein binding processes. By equilibrium and nonequilibrium molecular dynamics simulations, we show that importin-b, an archetypical a-solenoid, displays unprecedentedly large and fully reversible elasticity. Our stretching molecular dynamics simulations reveal full elasticity over up to twofold end-to-end extensions compared to its bound state. Despite the absence of any long-range intramolecular contacts, the protein can return to its equilibrium structure to within 3 A˚ backbone RMSD after the release of mechanical stress. We find that this extreme degree of flexibility is based on an unusually flexible hydrophobic core that differs substantially from that of structurally similar but more rigid globular proteins. In that respect, the core of importin-b resembles molten globules. The elastic behavior is dominated by nonpolar interactions between HEAT repeats, combined with conformational entropic effects. Our results suggest that a-sole- noid structures such as importin-b may bridge the molecular gap between completely structured and intrinsically disordered proteins. INTRODUCTION Solenoid proteins, consisting of repeating arrays of simple on repeat proteins focus on the folding and unfolding basic structural motifs, account for >5% of the genome of mechanism (14–16), an atomic force microscopy study on multicellular organisms (1). -

Investigation of Iron's Role in Alginate Production And
INVESTIGATION OF IRON’S ROLE IN ALGINATE PRODUCTION AND MUCOIDY BY PSEUDOMONAS AERUGINOSA by JACINTA ROSE SCHRAUFNAGEL B.S., University of Colorado Denver, 2003 M.S., University of Colorado Denver, 2006 A thesis submitted to the Faculty of the Graduate School of the University of Colorado in partial fulfillment of the requirements for the degree of Doctor of Philosophy Molecular Biology Program 2013 This thesis for the Doctor of Philosophy degree by Jacinta Rose Schraufnagel has been approved for the Molecular Biology Program by David Barton, Chair Michael Vasil, Advisor Mair Churchill Jerry Schaack Michael Schurr Martin Voskuil Date 10/30/13 ii Schraufnagel, Jacinta R. (Ph.D., Molecular Biology) Investigation of Iron’s Role in Alginate Production and Mucoidy by Pseudomonas aeruginosa. Thesis directed by Professor Michael Vasil. ABSTRACT Iron, an essential nutrient, influences the production of major virulence factors and the development of structured non-alginate biofilms in Pseudomonas aeruginosa. Fur, pyoverdine and various concentrations of iron sources have all been shown to affect non-alginate biofilm formation. Remarkably however, the role of iron in alginate biofilm formation and mucoidy is unknown. Because of alginate’s association with biofilms in chronic cystic fibrosis (CF) lung infections, we examined the influence of iron on alginate production and mucoidy in P. aeruginosa. Herein, we identify and characterize a novel iron-regulated protein that affects the transcription of alginate biosynthesis genes. Further, contrary to what is known about non-alginate biofilms, we discovered that biologically relevant concentrations of iron (10 µM – 300 µM), in various forms (e.g. FeCl3, Ferric Citrate, Heme), led to the decreased expression of alginate biosynthesis genes and the dispersion of alginate biofilms in mucoid P. -

Supporting Information
Supporting Information Barquilla et al. 10.1073/pnas.0802668105 SI Materials and Methods The purification of recombinant GST proteins using glutathione Bioinformatics, Plasmids Constructs, and Cell Lines. Searches for T. Sepharose 4B (Amersham) was performed according to the brucei TOR, raptor, AVO3, and FKBP12 orthologues were manufacturer’s instructions and the proteins were dialyzed performed using GeneDB (www.genedb.org) and NCBI Blast against PBS. (www.ncbi.nlm.nih.gov/BLAST/). Motif analyses were carried out using InterPro and Prosite. Antibodies. Mouse anti-TbFKBP12 monoclonal antibodies and GeneDB database accession numbers of these orthologues rabbit polyclonal anti-TOR antibodies were produced by inject- are: TbTOR1 (Tb10.6k15.2060), TbTOR2 (Tb927.4.420), ing the recombinant proteins into a BALB-C mouse or rabbit TbTOR-like 1 (Tb927.4.800), TbTOR-like 2 (Tb927.1.1930), using standard immunization protocols. Experiments involving TbRaptor (Tb11.03.0460), TbAVO3 (Tb927.8.3200), animals were approved by the National Animal Research Com- TbFKBP12 (Tb927.7.3420), and TbRaptor-like (Tb927.1.2190). mittee. Anti-TbFKBP12 antibodies were raised against the DNA fragments (400–700 bp) of the TbTOR1, TbTOR2, whole protein fused to GST, and hybridomes F12–3C7H11 and TbTOR-like 1, TbRaptor, TbAVO3, TbFKBP12, and TbRaptor- F12–7D10 H8 were selected by standard monoclonal selection like were amplified by PCR using pairs of specific primers (Table procedures as anti-TbFKBP12 monoclonal antibody producers. S1) and cloned into pGEM-T (Promega). All constructs were Anti-TbTOR antibodies were raised against nonhomologous sequenced to confirm that the subcloned DNA fragments had no TbTOR CЈ-terminal regions between the kinase domain and the mutations.